Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | J. Carlos Menéndez | + 8819 word(s) | 8819 | 2021-06-10 06:00:34 | | | |
| 2 | Vivi Li | Meta information modification | 8819 | 2021-06-10 10:33:15 | | | | |
| 3 | Vivi Li | Meta information modification | 8819 | 2021-06-11 09:58:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Menéndez, J.C.; Cores, �.; Piquero, M.; Villacampa, M.; León, R. NRF2 Regulation. Encyclopedia. Available online: https://encyclopedia.pub/entry/10715 (accessed on 09 August 2026).
Menéndez JC, Cores �, Piquero M, Villacampa M, León R. NRF2 Regulation. Encyclopedia. Available at: https://encyclopedia.pub/entry/10715. Accessed August 09, 2026.
Menéndez, J. Carlos, Ángel Cores, Marta Piquero, Mercedes Villacampa, Rafael León. "NRF2 Regulation" Encyclopedia, https://encyclopedia.pub/entry/10715 (accessed August 09, 2026).
Menéndez, J.C., Cores, �., Piquero, M., Villacampa, M., & León, R. (2021, June 10). NRF2 Regulation. In Encyclopedia. https://encyclopedia.pub/entry/10715
Menéndez, J. Carlos, et al. "NRF2 Regulation." Encyclopedia. Web. 10 June, 2021.
Copy Citation
NRF2 acts by controlling gene expression, being the master regulator of the Phase II antioxidant response, and also being key to the control of neuroinflammation. NRF2 activity is regulated at several levels, including protein degradation by the proteasome, transcription, and post-transcription.
NRF2–ARE pathway
neurodegenerative diseases
oxidative stress
KEAP1
NRF2 regulation
1. The NRF2–ARE Pathway
After the discovery, in 1994, of the NRF2 transcription factor [1], its involvement in the regulation of the expression of about 250 genes containing an enhancer sequence called antioxidant response element (ARE) was shown [2]. These genes encode several enzymes involved in cellular protection against pro-oxidants, electrophiles or inflammatory agents, biotransformation reactions of xenobiotics, maintenance of mitochondrial function, protein homeostasis, and antioxidative metabolism [3].
The maintenance of NRF2 levels requires a suitable balance between its synthesis and its proteasomal degradation [4][5]. As summarized in Figure 1, under physiological conditions NRF2 is normally located in the cytosol [4]. KEAP1 (Kelch-like ECH-associated protein 1), the NRF2 main negative regulatory system [4], is able to maintain NRF2 in the cytoplasm to generate a complex with the Cullin 3 (Cul3)/Rbx1-based E3-ubiquitin ligase, inducing NRF2 ubiquitination and the subsequent proteasomal degradation [5]. Thus, NRF2 is constitutively synthesized and rapidly degraded by the proteasome under unstressed conditions [5]. However, in response to oxidative stress, the cysteine residues present in the “sensor” region of KEAP1 are oxidized, leading to a conformational change that releases NRF2 from KEAP1. Alternatively, these cysteine residues can be activated by reaction with electrophiles [6]. Once NRF2 is liberated from KEAP1, it translocates into the nucleus, where it forms heterodimers with different coactivators, such as the sMaf (small masculoaponeurotic fibrosarcoma) protein [7][6], and these heterodimers promote the transcription of ARE. Due to its cytoprotective effects, NRF2 activation stands out as a promising goal to fight neurodegeneration processes, which involve mitochondrial disfunction, chronic inflammation, and increased oxidative stress levels [8].
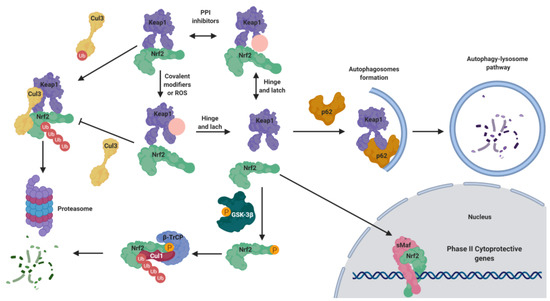
Figure 1. Negative regulation of nuclear factor (erythroid-derived 2)–like 2 (NRF2) under normal conditions and activation under pathological conditions. Under normal conditions, NRF2 is bound to Kelch-like ECH-associated protein 1 (KEAP1), maintaining it in the cytosol. KEAP1 acts as adaptor of the Cullin 3 (CuI3) ubiquitin ligase that ubiquitinates NRF2, leading to its degradation by proteasome 26S. Under pathological conditions, in the presence of reactive oxygen species (ROS) or covalent modifiers, the KEAP1 structure is modified, liberating NRF2 in the cytosol; thereafter, it translocates to the nucleus where it forms heterodimers with small masculoaponeurotic fibrosarcoma (sMaf) proteins to bind the antioxidant response element (ARE) sequences, promoting the expression of phase II genes. This mechanism is known as the “hinge and latch” NRF2 activation mechanism. Moreover, NRF2 is tightly regulated by additional mechanisms. It can be over-activated by the action of p62, since it can induce KEAP1 degradation by the autophagy lysosome pathway. In the opposite, under pathological conditions, in which the kinase glycogen synthase kinase-3β (GSK-3β) is over-active, it can directly phosphorylate NRF2, facilitating its interaction with the Cul1 ligase adaptor β-transducing repeat-containing protein (β-TrCp) to induce its degradation.
2. Structure of NRF2
NRF2 belongs to the basic-region leucine zipper (bZIP) transcription factors, and more specifically to the cap ‘‘n’’ collar (CNC) subfamily [9]. It is formed by seven NRF2–ECH homology domains known as Neh1–7, each with a specific function (Figure 2) [10]. The Neh1 domain contains the CNC–bZIP region that allows NRF2 to recognize DNA, promoting its heterodimerization with partners such as the sMaf protein [11]. The high degree of conservation found in the amino acid sequence of this region across a wide range of species [12] highlights the importance of NRF2 transcriptional activity. Neh2 is responsible for the interaction with KEAP1, the main NRF2 negative regulator [13]. This interaction takes place through the degron motifs present in this domain, namely DLG (low affinity) and ETGE (high affinity) [13]. Moreover, the presence of seven lysine residues, also included in the Neh2 region, promotes NRF2 proteasomal degradation after its ubiquitylation [6]. The C-terminal Neh3, Neh4, and Neh5 are transactivation domains that bind to various components of the transcriptional machinery, which promotes the transcription of NRF2 target genes [14][15]. Neh6 is a serine-rich region containing two conserved peptide motifs, DSGIS and DSAPGS [16]. Glycogen synthase kinase-3β (GSK-3β) is able to phosphorylate the DSGIS sequence, thereby increasing the binding efficiency of β-transducing repeat-containing protein (β-TrCP) to Neh6 and promoting NRF2 proteasomal degradation [16]. Finally, Neh7 is responsible for the binding with RXRα (Retinoic acid receptor-alpha) and disrupts the binding of CBP ([CREB (cAMP-response-element-binding protein)-binding protein]) to the Neh4 and Neh5 domains, inhibiting the transcription of the ARE sequences [17].
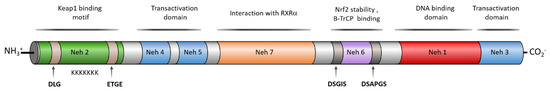
Figure 2. Human NRF2 domain structure and the activity associated to each domain. NRF2 is composed by seven Neh domains with different interaction patterns and specific activities. Neh1 presents a bZip region involved in dimerization with sMaf proteins to bind ARE regions and other transcription factors such as c-JUN, Sp-1, and JDP2 (Jun dimerization protein 2). Neh1 is also an acetylation-sensitive domain, contains an NES sequence to be exported by Crm1 and it is the target of phosphorylation by AMPK (AMP-activated protein kinase). Neh2 is a domain for degradation (degron domain) that presents the binding domain of KEAP1 and other ubiquitin ligases. KEAP1 targets the DLG and ETGE motifs each of them with different affinity. Neh 2 contains a α-helix composed by 7 Lys residues that are the targets for ubiquitination, an NLS sequence, and a serine residue (S40), that is a phosphorylation target of PKCδ, and both motifs are related to NRF2 nuclear translocation. The Neh3 domain contains another NLS sequence and two Lys residues that are targets for acetylation related to nuclear translocation. Neh4 and Neh5 are transactivation domains involucrate in the binding of CBP, P300, Hrd1, and RAC3 to NRF2. Neh6 contains a second degron domain targeted by β-transducing repeat-containing protein (β-TrCP) by interaction with the DSGIS motif, after being phosphorylated by GSK-3β and the DSAPGS motif. Finally, Neh7 is a repressor domain targeted by RXRα.
The classical NRF2 target genes encode enzymes distributed in several organelles and subcellular compartments [18]. These enzymes participate in metabolic reactions that scavenge ROS and neutralize electrophiles such as superoxide dismutase (SOD), glutathione peroxidase, catalase, glutathione reductase (GR), glutamate cysteine ligase (GCL), or NAD(P)H/quinone oxidoreductase 1 (NQO1) [18]. NRF2 also plays a key role in the induction of genes involved in drug metabolism and distribution, including genes encoding non-cytochrome P450 phase-I and phase-II drug metabolism enzymes [19]. Furthermore, it promotes the degradation of the heme group by inducing heme oxygenase-1 (HO-1), and some of the induced enzymes are involved in the generation of antioxidant small molecules such as glutathione synthase, which is essential for the biosynthesis of glutathione (GSH), the most abundant antioxidant and electrophile-neutralizing molecule inside the cells [20]. NRF2 also regulates proteins related to key processes such as autophagy [21], promotes the preservation of mitochondrial functions [22], and inhibits the expression of proinflammatory cytokines [23].
3. NRF2 Activation as a Useful Approach to Neurodegenerative Disease Therapy
NRF2 deficiency has been found in several NDDs; for instance, the hippocampal neurons of AD patients showed a dramatic decrease in nuclear NRF2 [24], while animal models of PD, such as NRF2-knockout mice, showed a specific loss of dopaminergic neurons [25] and post-mortem studies of patients with ALS showed an increased KEAP1 mRNA in the motor cortex [26], leading to a decline in NRF2 activity. Other studies showed that tau- and amyloid-injured neurons contain increased levels of NRF2 and its target protein sequestosome 1 (SQSTM1/p62), probably as a clearance mechanism to release these toxic proteins through autophagy [21][27]. In consonance with these results, the levels of HO-1, NQO1, GCLM, and SQSTM1 levels are increased in AD and PD brains [27][28]. In this context, it is not surprising that NRF2 activation has been demonstrated to extend survival in AD [29], PD [30], and ALS [31] animal models.
The low levels in the expression or activity of NRF2 in neurodegeneration models together with the decrease of the neurodegenerative process upon NRF2 induction indicate that the NRF2–ARE pathway is a promising target for NDDs [32][33]. Furthermore, this pathway regulates multiple pathological processes implicated in neurodegeneration processes such as oxidative stress, neuroinflammation, and aberrant proteostasis, by targeting NRF2, several pathways can be simultaneously modulated [21][23][32][34].
3.1. KEAP1-Dependent Regulation
NRF2 regulation is accomplished by several cellular factors controlling its stability and nuclear translocation, but among them, the previously mentioned KEAP1 protein is the most important [6].
3.1.1. KEAP1 as an Oxidative Stress Sensor
KEAP1 is a Zn metalloprotein with 625 amino acid residues and it has a branched stem dimeric structure, formed by 5 domains [35]. The N-terminal region (NTR), the BTB/POZ (Bric-a-brac, tramtrac, broad-complex/proxvirus zinc fingers) domain, implicated in KEAP1 homodimerization and Cul3 association, the intervening region (IVR), which acts as a linker between BTB and DGR domains, the double glycine repeat (DGR) domain, important for NRF2 regulation and actin interaction and the C-terminal domain (CTR) [35] (Figure 3).
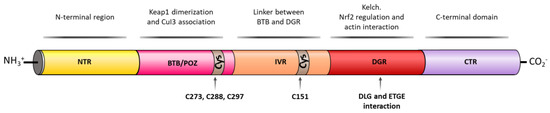
Figure 3. KEAP1 structural domains and their related activity. KEAP1 contains an N-terminal region (NTR), a Tramtrack and Bric-á-Brac (BTB) domain that enables KEAP1 homodimerization and responsible for Cul3 E3 ligase binding. The BTB domain also contains three important Cys residues (Cys273, Cys288, and Cys297) able to react with free radicals and/or electrophiles inducing a conformational change in KEAP1 that liberates NRF2 in the cytosol. The next domain is an intervening region (IVR) that also contains an important Cys residue, Cys151, which is able to react with free radicals and or electrophiles and an NES motif that regulates the cytoplasmatic location of KEAP1. The double glycine repeat (DGR) domain contains six kelch repeats that contain the binding sites of NRF2, p62, and related E/STGE proteins. Finally, KEAP1 contains the C-terminal domain (CTR).
KEAP1 is a cysteine-rich protein, being redox-active and reactive to the local environment [36]. The majority of KEAP1 cysteine residues are surrounded by basic amino acids that increase their reactivity by lowering their pKa values [37]. Thus, the 27 cysteine residues of KEAP1 are proposed to be the main sites through which the protein is able to sense electrophilic or oxidative stress [38]. Even though cysteines are distributed along the whole KEAP1 sequence, it has been proved that the C273, C288, and C297 residues in the BTB/POZ domain [39] and C151 in the IVR domain [40] are more reactive and serve as redox sensors for electrophiles, reactive oxygen species (ROS), and metals such as Cd2+, As3+, and Se4+ [41]. The chemical modification of their sulfhydryl groups decreases NRF2 ubiquitination, causes KEAP1–NRF2 dissociation, and promotes the nuclear translocation of NRF2 [42].
The ‘hinge and latch’ model [43] is the most widely accepted mechanism for KEAP1-dependent regulation. In this model, two different binding sites of NRF2 in the Neh2 domain interact with different affinities with a single overlapping site in the DGR domain of KEAP1, with the ETGE motif presenting higher affinity than DLG [44]. Under homeostatic conditions, the β-hairpin of the ETGE motif (high affinity) interacts with one of the KEAP1 subunits in a first step, which is called the open conformation [44][45]. This open conformation represents the “hinge”, in which NRF2 can move in space relatively freely. In a second step, the β-hairpin of the DLG motif (low affinity) interacts similarly with a second protomer of KEAP1 in the so-called closed conformation, representing the “latch” [44][45]. This closed conformation restricts the ability of NRF2 to move and enables an optimal positioning of a Lys-rich α-helix present in the Neh2 region of NRF2; this helix is the target for ubiquitin conjugation, consequently promoting NRF2 for proteasomal degradation [45].
Under oxidative stress conditions, NRF2 inducers oxidize the Cys residues present in KEAP1, promoting a conformational change [45]. This structural change in KEAP1 causes an improper spatial disposition of the target lysines, and thus NRF2 is no longer ubiquitinated nor degraded [46]. This leads to the saturation of KEAP1, and consequently any newly synthesized NRF2 can evade repression and directly accumulate into the nucleus, promoting its target gene expression [47].
3.1.2. KEAP1 Covalent Modifiers
While NRF2 can be physiologically activated by an increase of oxidative stress, it can also be exogenously induced by chemical agents [44]. The majority of known NRF2 inducers are electrophiles that modify the cysteine residues present in the thiol-rich domain of KEAP1 [48][49] in a covalent way, either by oxidation or alkylation. These electrophile adducts can inhibit KEAP1 in two different ways, namely: (1) by promoting a conformational change in KEAP1 that will inhibit his binding with NRF2, or (2) by blocking the interaction between KEAP1 and Cul3/Rbx1, inhibiting NRF2 ubiquitination and promoting its sequestration and the subsequent stabilization of the newly synthetized NRF2 [50][51].
In the past years, many natural and synthetic compounds have been described to potentially activate the NRF2 pathway through this mechanism, and these are summarized below.
Natural and Semisynthetic NRF2 Activators and Their Analogues
Caffeic acid (2, Figure 4) is a polyphenol present in coffee that bears a Michael acceptor embedded in its structure. Some studies reveal that this moiety is responsible and essential for its NRF2 induction properties while its nucleophilic functional groups (catechol) provide clearance but are not directly involved in NRF2 induction [52]. Further studies have shown that caffeic acid is also able to decrease KEAP1 expression to activate NRF2, leading to an increase in the expression of HO-1 (heme oxygenase 1) and NQO1 (NAD(P)H:quinone oxidoreductase 1) [52].
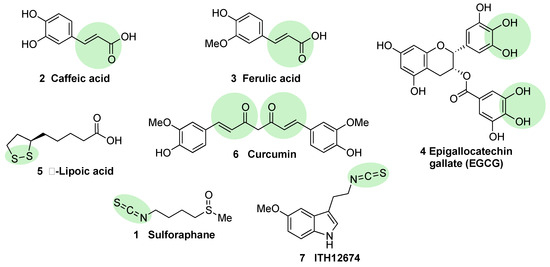
Figure 4. Chemical structures of natural NRF2 activators and their analogues. Caffeic acid (2), ferulic acid (3), and curcumin (6) are considered classical Michael acceptors where a double bound is conjugated with a carbonyl group, creating a potent electrophile. Epigallocatechin gallate (4) is a polyphenolic compound that is able to generate quinone derivatives after oxidation, and the corresponding quinone exerts a potent electrophilic character. Lipoic acid (5) is considered an electrophile that is able to react with KEAP1 Cys residues to form lipoylcysteinyl disulfides, thus modifying their structure. Finally, isothiothianate moieties at sulforaphane (1) and its hybrid melatonin derivative, ITH12674 (7), are responsible for the potent electrophilic character of these compounds. The electrophilic moieties or promoieties of each compound are highlighted in green.
Ferulic acid (3, Figure 4) is widespread in vegetables, and its main structural difference with caffeic acid is one methoxy group on its benzene ring. It has been proved that ferulic acid is able to regulate the NRF2 pathway and counteract trimethyltin (TMT)-induced neuronal damage in the human neuroblastoma cell line SH-SY5Y [52]. Moreover, it is able to antagonize oxidative stress by ROS scavenging and activating the non-homologous end-joining DNA repair process [52].
Epigallocatechin gallate (EGCG) (4, Figure 4), a catechin found in green tea [53], upregulates the NRF2 pathway through electrophilic disruption via its prior auto-oxidation of its catechol moieties to ortho-quinones [54], as well as via activation of the p38-MAPK and extracellular signal-regulated kinases (ERK)1/2 signaling pathways [55]. EGCG has shown multiple in vivo and in vitro neuroprotective effects in different models of MS, PD, and traumatic brain injury, all of them associated with an increase in NRF2 induction, antioxidant activity, and a decrease of inflammatory responses [56][57][58].
Alpha lipoic acid (5, Figure 4) can be found in a wide number of plants including carrots, beets, broccoli, and spinach. The exact mechanism through which it induces NRF2 is not currently known, although in view of its structure, it can form lipoylcysteinyl disulfides with KEAP1, preventing NRF2 degradation [59][60]. Besides this potential mechanism, it has also been shown that alpha lipoic acid can activate protein kinase C (PKC), which is one of the kinases that is able to activate NRF2 through an alternative pathway [61]. Several studies on alpha lipoic acid neuroprotective effects have been carried out, although in many of them, it was not investigated whether NRF2 activation took part in these effects. In mice MS models, alpha lipoic acid reduced inflammation [62]. In addition, in PD models, it decreased ROS, restored ATP levels, preserved dopaminergic neurons, and upregulated mitochondrial formation [63].
Curcumin (6, Figure 4) is the main curcuminoid present in turmeric acid; it has potent antioxidant and anti-inflammatory properties [64][65]. It activates NRF2 by electrophilic modification of Cys-151 in KEAP1, modifying also its ROS-scavenging activity [66]. On the other hand, curcumin is also able to activate NRF2 by repressing KEAP1 expression [67]. Curcumin showed beneficial neuroprotective activities in intracerebral hemorrhage as well as in traumatic brain injury models [23][68]. A decreased in proinflammatory gene expression through the prevention of NF-kB activation in microglial cells was also observed upon treatment with curcumin [69].
Sulforaphane (1, Figure 4) is an isothiocyanate produced from the enzymatic cleavage of the organosulfur compound glucoraphanin found in cruciferous plants such as broccoli, brussels sprouts, cauliflower, and cabbage [7]. The catalytic reaction needed for the release of sulforaphane is driven by a β-thioglucosidase present in the gut microbiota [70]; thus, the levels of released sulforaphane are highly dependent on dietary habits and microbiome composition [70]. Sulforaphane directly interacts with the Cys151 residue in KEAP1, thereby promoting NRF2 activation [71][72]. Either in pure form or as sprout extract, sulforaphane has shown a very good safety profile, and more than 30 clinical studies against chronic diseases have been carried out [73][74]. It has the ability to cross the blood–brain barrier (BBB), and it has shown neuroprotective capacity against Aβ1-42 peptide in neuronal cells [75]. In vivo, sulforaphane alleviates cognitive impairment in mouse models of AD [76]. In PD models, it protected dopaminergic neurons against the parkinsonian toxin 6-hydroxydopamine [77]. Furthermore, sulforaphane is able to reduce the levels of phosphorylated tau and increase Beclin1 and LC3-II, leading to the conclusion that NRF2 activation may facilitate tau degradation through autophagy [78]. Nevertheless, sulforaphane is an oily substance with low stability in hydrophilic media, and its pharmacokinetic properties need to be improved [7]. In this regard, sulforadex (SFX-01), a sulforaphane-β–cyclodextrin complex, has shown an excellent bioavailability and is under clinical trials for the treatment of subarachnoid hemorrhage and metastatic breast cancer [7]. We will finally mention that sulforaphane has been hybridized with the natural antioxidant melatonin generating ITH12674 (7, Figure 3), which is a compound designed to have a dual drug–prodrug mechanism of action for the treatment of brain ischemia [79].
Semisynthetic cyanoenone triterpenoids derived from bardoxolone (RTA401, CDDO, 8a, Figure 5), described as antioxidant inflammation modulators (AIMs), exhibit a Michael acceptor moiety and are among the most potent known electrophilic NRF2 activators [80], which interact with Cys151 in KEAP1 and are under clinical development by Reata Pharmaceuticals and Kyowa Hakko Kirin. Proof-of-concept studies strongly support the use of these triterpenoids for degenerative diseases [80]. For instance, CDDO-ethyl amide (RTA405, 8b, Figure 5) and CDDO-trifluoethyl amide (RTA404, 8c, Figure 5) induced a significant reduction of toxicity levels in PD models [30]. CDDO-methyl ester (CDDO-Me, RTA402, 8d, Figure 5) was the first CDDO to reach clinical trials for the treatment of diabetic nephropathy, although it was later withdrawn at phase III (BEACON trial) due to cardiovascular safety issues that are not related with NRF2 induction [81]. In an effort to improve the safety profile, CDDO-difluoropropionamide (RTA408, omaveloxone, 8e, Figure 5) was synthetized and is currently in phase II trial for the treatment of FRDA, ocular inflammation, and pain after ocular surgery [82].
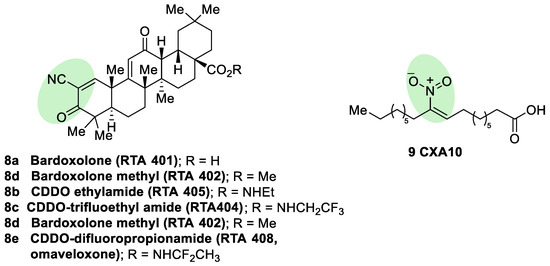
Figure 5. Chemical structures of some semisynthetic cyanoenone triterpenoids (8) and a nitro fatty acid (9). These compounds are considered electrophilic NRF2 inducers due to the presence of α,β-unsaturations conjugated with carbonyl, cyano, or nitro groups. These electrophilic moieties are highlighted in green.
Nitro fatty acids (NO2-FAs) are endogenous signaling mediators with anti-inflammatory and antifibrotic activities in preclinical animal models of metabolic and inflammatory disease [83]. The nitro alkene structure confers electrophilicity to their β-carbon, enhancing the formation of Michael adducts with nucleophiles such as cysteines. It has been proven that NO2-FAs react with Cys-273 and Cys-288 of KEAP1, activating NRF2 [84]. The reversibility of this reaction [85] prevents the formation of stable adducts, which could lead to toxicity. Furthermore, compound 9 (10-NO2-OA, CXA10, Figure 5) proved to be safe in humans at an active dose in a phase I safety assay [86], and it is currently being tested for the treatment of focal segmental glomerulosclerosis.
Synthetic NRF2 Activators
Fumaric acid esters are the group of synthetic NRF2 activators that are receiving the highest amount of attention from the pharmaceutical industry [18]. Dimethyl fumarate (DMF) (10, Figure 6) is the most clinically successful member of this family. It was firstly approved in 1994 for the treatment of psoriasis [87], but due to its efficacy in multiple sclerosis mouse models, it was repurposed in 2013 by Biogen under the name Tecfidera for the treatment of relapsing-remitting MS [88], and it has become one of the most successful new medicines in the past few years [7]. Due to its thiol-reactive Michael acceptor structure, DMF primarily activates NRF2 via cysteine modification on KEAP1 [89], and it has also been proven to affect NRF2 phosphorylation via the phosphatidylinositol-3-kinase (PI3K) and ERK1/2 pathways [90].
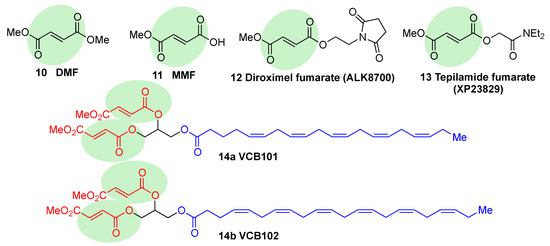
Figure 6. Chemical structures of fumaric acid ester derivatives. Their electrophilic moieties are highlighted in green.
DMF has shown to be protective in a wide range of neurodegenerative conditions in an AD model [91] and to prevent hippocampal injury after ischemia and protect BBB integrity in mouse models of stroke [92]. Furthermore, protection against α−Syn and Aβ toxicity and a reduction in tau hyperphosphorylation [93][94][95] was also observed.
DMF is mostly converted into monomethyl fumarate (MMF, 11, Figure 6) by intestinal esterases. Moreover, it has been shown that MMF reacts with Cys151 in KEAP1, thereby activating NRF2 [96]. Several biopharmaceutical companies are developing slow-release forms of MMF in order to improve its bioavailability and reduce the side effects associated to DMF [80]. Thus, Alkernes has developed ALK8700 (diroximel fumarate, 12, Figure 6), an MMF prodrug with reduced side effects that is now in phase III trials for MS [97]. Tepilamide fumarate (XP23829, 13, Figure 6) is another MMF prodrug, which has been developed by XenoPort [18]. This compound has shown better solubility and permeability compared to DMF, and also an improved efficacy and reduced gastrointestinal side effects, and it is now in phase II clinical trials for the treatment of plaque psoriasis [18]. A conjugate of MMF and docosahexaenoic acid (CAT4001) is being developed by Catabasis Pharmaceuticals. In animal models, it has shown promising activities for the treatment of NDDs, such as FRDA and ALS [18]. With a similar design, V ClinBio developed conjugates built around central glycerol unit and containing two molecules of MMF linked to one of eicosapentenoic acid (VCB101, 14a, Figure 6) or docosahexenoic acid (VCB102, 14b, Figure 6) for the treatment of MS and psoriasis [18].
OT551 (15, Figure 7) is an N,N-disubstituted hydroxylamine developed by Othera Pharmaceuticals [7][98]. It is a prodrug whose active metabolite TEMPOL (4-hydroxy TEMPO, 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl) inhibits oxidative stress and disease-associated inflammation by a complex mechanism that, besides its radical scavenging activity, involves a reduced activation of NF-κB [99] in acute inflammation and activation of the KEAP1–NRF2 pathway, both indirectly and by targeting KEAP1 [100]. It was designed for topical use and protected the retinal pigment epithelium and photoreceptors from oxidative damage and inflammation in preclinical studies. It has also shown efficacy in a phase II clinical trial on age-related macular degeneration [18].
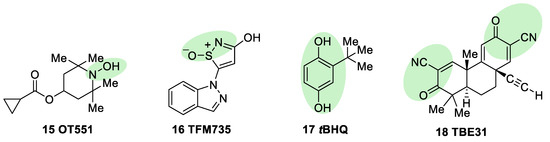
Figure 7. Chemical structures of OT551, TFM735, tBHQ, and TBE31. Their electrophilic moieties or promoieties are highlighted in green.
During a high-throughput screening study, TFM735 (16, Figure 7) was identified as an activator of NRF2 through a Cys151-dependent mechanism [101]. This compound is currently in preclinical development by Mochida Pharmaceuticals for the treatment of MS [18].
tBHQ, tert-butylhydroquinone (17, Figure 7) is a metabolic precursor of an electrophilic quinone that is able to disrupt the KEAP1/NRF2 complex. It is widely used as a food preservative and due to its antioxidant properties, it has been also employed with neuroprotective purposes. Treatment with tBHQ reduced oxidative stress, and it also prevented neuronal toxicity and Aβ formation in NT2N cell lines [102]. Furthermore, it also led to a decrease in secondary injury and an improvement in function recovery after traumatic brain injury [103].
TBE-31 (18, Figure 7), a fully synthetic acetylenic tricyclic bis(cyanoenone), is one of the most potent known NRF2 inducers [59]. It has been mainly studied in cancer models, presenting strong antioxidant and anti-inflammatory activities. Furthermore, treatment with TBE-31 improved mitochondrial function and protection against oxidative stress in fibroblasts and cerebellar granule neurons in FRDA models [104].
3.1.3. KEAP1 Protein–Protein Interaction Inhibitors
KEAP1 is the main regulator of NRF2, by promoting its degradation [5]. The NRF2 recognition by the Kelch domain included in KEAP1 is a classic example of a protein–protein interaction as a drug target, which, generally speaking, remains a challenge for drug discovery due to the large surface of the targeted area and the lack of pockets for the interaction with inhibitors. However, in the case of KEAP1, the target surface is about 300–1000 Å2 in size and has well-defined pockets with residues capable of stably recognizing small molecules [105]. These topological characteristics, together with the availability of KEAP1 3D structures, allow the development of small molecules that are able to recognize KEAP1 and disrupt its binding with other proteins [106].
The ETGE motif contained in the Neh2 domain of NRF2 adopts a β-hairpin conformation to interact with the KEAP1 Kelch domain-binding pocket, which can be artificially subdivided into five subpockets (Figure 8a) [106]. The P1 and P2 subpockets are characterized by the presence of multiple positively charged arginine residues (Arg415, Arg483, and Arg485), enabling these sites to recognize negatively charged amino acid residues of substrates [107]. In particular, the carboxyl side chains of Glu79 and Glu82 at the ETGE motif of NRF2 (Figure 8b) promote binding by their interaction with the P1 and P2 subpockets. The central P3 subpocket is formed by small polar and nonpolar residues that stabilize the ETGE motif. The external P4 and P5 subpockets are formed by aliphatic chain and tyrosine-rich residues and improve the substrate recognition through hydrophobic interactions and hydrogen bonds [106][107].
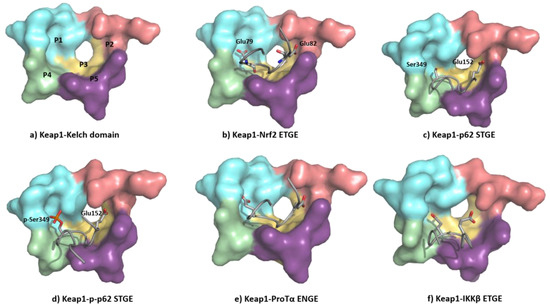
Figure 8. (a) Structure of the Kelch domain of KEAP1 (PDB: 5WTV), highlighting binding pockets P1: blue (residues 415, 461, 462, 478, 483 and 508); P2: red (363, 380, 381 and 414); P3: yellow (364, 509, 556, 571, 602 and 603); P4: green (334, 572 and 577); P5: purple (525, 530 and 555); (b) structural detail of the NRF2 ETGE motif bound to KEAP1 (PDB: 5WTV); (c) structural detail of the p62 STGE motif (PDB: 3ADE). The STGE motif in the KEAP1-interacting region (KIR) is similar to the ETGE-like motif of NRF2 and they both adopt similar β-turn conformations. However, while Glu79 in ETGE interacts with the Arg415 contained in the KEAP1 P1 subpocket, the corresponding Ser349 in STGE is too short, leading to a poor binding affinity; (d) structural detail of the Ser349-phosphorylated p62 STGE motif (PDB: 3WDZ). Phosphorylated Ser439 is deeply inserted into the P1 subpocket, and the negative charges increase the affinity through electrostatic interactions with Arg415 and Arg483; (e) structural detail of Protα ENGE motif (PDB 2Z32); (f) structural detail of the IKKβ ETGE motif obtained by molecular dynamics [108]. The substitution of the Thr residue (DEETGE, NRF2) by an Asn residue (NEENGE, ProTα) renders the β-turn conformation unstable, leading to a less potent binding between the ENGE motif and KEAP1 compared with that of the ETGE motif in NRF2.
The Kelch domain in KEAP1 is able to interact with several additional proteins containing ETGE-like sequences in a similar manner to NRF2 [109]. Some of them, such as SQSTM1/p62 and prothymosin α (ProTα), are also related to the regulation or the NRF2–ARE pathway, or they are implicated in the crosstalk with other hallmarks of neurodegeneration, such as IKK-β (inhibitor of the nuclear factor kappa-B kinase subunit beta) in neuroinflammation [109].
The KEAP1 Kelch domain recognizes p62 through a KEAP1-interacting region (KIR) and this disrupts the KEAP1–NRF2 system, leading to a crosstalk between the NRF2–ARE pathway and p62 [110] (Figure 8c). The mTORC1-dependent phosphorylation of this Ser349 elongates the side chain and adds negative charges to it, improving its recognition by KEAP1 (Figure 8d) [111]. The p62 protein is a cargo receptor for selective autophagy, and its strong interaction with KEAP1 allows it to sequester the latter into early autophagosomes, promoting its degradation through the autophagy–lysosome pathway [112]. Both the p62-mediated disruption of the KEAP1–NRF2 interaction and the p62-dependent degradation of KEAP1 increase the stability of NRF2, leading to neuroprotection [113]. Another ETGE-like substrate that can bind KEAP1 is ProTα, whose ENGE motif mediates its interaction with the KEAP1 Kelch domain (Figure 8e). The crystal structure of the KEAP1–ProTα complex shows a similar network of interactions to the NRF2 ETGE motif [114]. ProTα exhibits a neuroprotective profile against ischemia–reperfusion injury, which can be explained by its capacity to disrupt the NRF2–KEAP1 complex when present in a large excess [115][116].
KEAP1 also interacts with IKKβ, which plays a central role in the control of the pro-inflammatory NF-κB pathway after phosphorylation and several downstream steps [117]. The recognition between KEAP1 and IKKβ promotes the degradation of the latter and prevents the phosphorylation of IKKβ, reducing neuroinflammation [118]. The interplay between the NRF2–ARE and NF-ĸB pathways seems especially interesting in neurodegenerative and cerebrovascular diseases and can be explained in part by the interaction of KEAP1 with NRF2 and IKKβ [118]. Although a crystal structure of the KEAP1–IKKβ complex is unavailable [119], it has been studied through protein–protein docking and molecular dynamic analysis, which show that the ETGE motif of IKKβ adopts a β-turn conformation [119], occupying the substrate-binding pocket in a similar fashion to that of the NRF2 ETGE motif (Figure 8f) [108].
Small peptides based on structural similarity to KEAP1 substrates were the earliest examples of molecules designed to act as KEAP1 inhibitors [120]. However, they have several limitations, including their inability to cross the BBB and their poor stability. Thus, the development of small molecules as KEAP1 binding disruptors remains the main approach, overcoming the bioavailability and stability issues associated to peptides [121]. HTS of a library of more than 300,000 compounds led to tetrahydroisoquinoline 19 (Figure 9) as the first-in-class small-molecule KEAP1–NRF2 PPI inhibitor [121]. Further research efforts on this scaffold led to new structure–activity relationships [122]. The naphthalene sulfonamide derivative 20a (Figure 9) was also identified as a KEAP1–NRF2 inhibitor in a fluorescence anisotropy assay [123]. Structural determination showed an interaction between 20a and the P3–P5 subpockets in the Kelch domain of KEAP1. Based on this finding, compound 20b (Figure 9) was designed [123] with its two carboxyl groups forming multiple hydrogen bonds and electrostatic interactions with the basic Arg residues present in P1 and P2, thus improving the affinity with the KEAP1–NRF2 interface [123]. In the development process, the naphthalene core was replaced by an isoquinoline core to give 20c (Figure 9), improving the solubility, metabolic stability, and reducing the intrinsic toxicity of naphthalene scaffold [124]. Recently, the exploration of bioisosteric replacements of both carboxylic acids afforded the ditetrazole analogue 20d (Figure 9), which maintains the potent PPI inhibition activity and improves the cellular potency thanks to a better pharmacodynamic profile [125]. Another study demonstrated that oxadiazole-urea based compounds 21a-c (NK-252, Figure 9) interact with the KEAP1–Kelch domain [126], with the carboxylic group and the urea–oxadiazole structure interacting via hydrogen bonds and the phenyl moiety by π-stacking with the P1, P2 and P3 subpockets, respectively.
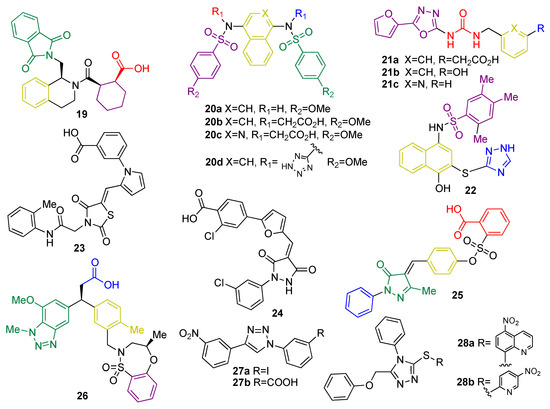
Figure 9. Chemical structures of KEAP1 protein–protein interaction inhibitors 19–28. Compounds 19, 20, 21, 22, 25 and 28 have been crystalized with KEAP1 and/or their interaction pattern has been proposed by using molecular modeling and molecular dynamics. Their interaction pattern with KEAP1 has been coded by giving different colors to the motif of each molecule that interacts with each pocket as follows: interaction at P1: blue; interaction at P2: red; interaction at P3: yellow; interaction at P4: green; and interaction at P5: purple.
Virtual screening of a 300,000-compound library afforded several hit structures that are able to interact with the KEAP1–NRF2 interface, including compounds 22, 23, and 24 [127]. Compound 25, structurally related to 20, exhibits a very interesting profile as a neuroprotector in a PD animal model by inhibiting the KEAP1–NRF2 interaction [128]. Fragment-based drug discovery was successfully employed in the development of 26, which is a novel phenylpropanoic acid-based KEAP1–NRF2 PPI inhibitor [129]. Crystallographic studies revealed the overlapping between the original fragments and 26 in the binding pocket [129]. This compound showed potency in cell-based assays in the nM range and was shown to activate the NRF2 pathway in vivo [67]. Similarly, simpler five-membered heterocycles such as 1,4-diphenyl-1,2,3-triazoles 27a-b were designed in silico to mimic the Glu residues in NRF2 and were shown to interfere in the KEAP1–NRF2 recognition process and induce the expression of NRF2 target genes in live cells [130]. Isomeric compounds containing the 1-phenyl-1,3,4-triazole scaffold were also described in the patent literature [131]. Compounds 28a and 28b (Figure 9) inhibit the KEAP1–NRF2 interaction with moderate inhibitory potency and induce the expression of NRF2 downstream target genes, leading to neuroprotection in an in vitro model of PD [131]. In particular, compounds 28 are particularly promising as anti-neurodegenerative agents, since in vivo studies using a HD model revealed that they inhibit the neurodegeneration of medium spiny neurons [132].
Research into KEAP1–NRF2 PPI inhibition is rapidly becoming a hot topic in medicinal chemistry, and it is evolving rapidly. In this context, the availability of crystal structures and a broad range of structural analyses related to recognition processes at the KEAP1–NRF2 complex have contributed to the development of several lead compounds by modern drug discovery strategies and the exploration of new chemical sources [122][127][131]. However, these molecules are often polar and possess a relatively large molecular weight, with a limited BBB penetration, leading to poor pharmacodynamic and pharmacokinetic profiles in NDDs [106]. Therefore, PPI inhibitor design with high potential activity and improved physicochemical properties is crucial to develop a new generation of drugs with a better safety profile, compared to KEAP1 covalent inhibitors.
3.1.4. Epigenetic Control of KEAP 1 Expression
Histone deacetylases (HDACs) remove acetyl groups in histones associated to the KEAP1 promoter region, inducing an increase in KEAP1 transcription [133]. Consequently, HDAC inhibitors, which counteract this effect, improve the redox balance and attenuate neuronal degeneration in some NDDs such as HD [133], ALS [134], and in animal models of stroke [135]. In a model of transient cerebral ischemia, treatment with suberolhydroxamic acid (SAHA) (29, Figure 10), also known as vorinostat, reduced infarct volume by 30–40% [136]. Trichostatin A (TSA) (30, Figure 9) was identified as an inhibitor of KEAP1 expression and leads to an improvement of NRF2 activity and protection against cerebral ischemia [137].
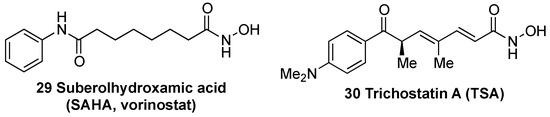
Figure 10. Chemical structures of histone deacetylases (HDAC) inhibitors suberolhydroxamic acid (SAHA) (29) and trichostatin A (TSA) (30).
MicroRNAs (miRNAs or miRs) are small noncoding RNAs with 18–25 nucleotides in length that are able to bind to the 3′-untranslated region (UTR) of the target mRNAs [138]. After miRs and target mRNA binding, and depending on the complementarity of both sequences, the target mRNA is degraded or its transcription is suppressed [138]. The miR-7 reduce the expression of KEAP1 after the interaction with its 3′-UTR in the SH-SY5Y cell line [139]. This downregulation of KEAP1 expression leads to NRF2 stabilization and increases the expression of several cytoprotective proteins. The effect of miR-7 appears to be especially interesting in PD models, where the overexpression of miR-7 protects neuronal cells against oxidative stress [139]. It is also relevant that the brain regions more affected in PD, such as substantia nigra and striatum, show an increased level of miR-7 compared with non-affected regions such as the cortex or cerebellum [140]. Furthermore, miR-7 expression downregulation observed in a PD mice model [140] suggests its implication in PD development.
Based on these precedents, KEAP1 expression regulation by epigenetics or miRs can provide an effective strategy to improve neuroprotection through the NRF2–ARE pathway.
3.2. KEAP1-Independent Regulation
As discussed above, NRF2 activation is mainly regulated by KEAP1, which is a mechanism known as the canonical pathway. However, many non-KEAP1-related processes contribute to NRF2 regulation and are summarized in this section.
3.2.1. NRF2 Regulation by GSK-3β
GSK-3β is a Ser/Thr kinase involved in glycogen metabolism, cell proliferation, apoptosis, and other functions within the cell [141]. Regarding NDDs, GSK-3β activity has been reported to be upregulated in AD and has been considered to play a key role on tau hyperphosphorylation [142]. Therefore, GSK-3β has been one of the most important targets in drug development toward AD. In 2006, Rojo et al. described the ability of this kinase to modulate the NRF2–ARE response [143], promoting NRF2 phosphorylation and nuclear exclusion mediated by Fyn [144]. GSK-3β phosphorylates Fyn, to increase its activity; in turn, Fyn phosphorylates NRF2 at Tyr568, leading to its extranucleation [145] and degradation.
Additionally, another line of research has demonstrated that GSK-3β phosphorylates the DSGIS [15] degron motif at Neh6 of NRF2 [146], as described above. This post-translational modification increases the affinity of β-TrCP for NRF2 to induce its proteasomal degradation [15], and it is further discussed in next paragraph. Moreover, GSK-3β its activity is regulated by several kinases: the PI3K/Akt pathway downregulates GSK-3β by the phosphorylation of Ser9 [147] to induce its inactivation [148], and p38-MAPK also regulates its activity by phosphorylation at Thr390 [149].
Several GSK-3β inhibitors have reached clinical trials toward different diseases including AD, mild cognitive impairment, autism spectrum disorders, and cancer. Initially, NRF2 activation via GSK-3β inhibition was demonstrated for LiCl, a GSK-3β inhibitor used for the treatment of bipolar disorder and compound TDZD-8 [150], which is able to increased HO-1 expression. Recently, LiCl-induced NRF2 activation has been reported in different in vitro and in vivo models, confirming the regulation of the latter by GSK-3β [151]. TDZD-8-induced NRF2 activation has been further demonstrated in an in vivo model of renal ischemia/reperfusion injury. It reduced oxidative stress and cellular apoptosis via NRF2 activation [152].
A clinically advanced example is tideglusib, a thiadiazolidinone derivative, which was developed as a non-ATP competitive GSK-3β inhibitor for AD treatment, which reached phase II clinical trial (ARGO study) [153], but it failed to produce clinical benefit. Researchers suggested a potential negative effect of the non-linear dose response and propose further dose-finding studies in early AD cases. Tideglusib and pioglitazone, also a thiadiazolidinone derivative, showed neuroprotection against MPPT toxicity mediated by NRF2 induction and phase II response activation. In this model, authors demonstrated that the activation of the antioxidant response was mediated by GSK-3β inhibition [154], although the neuroprotective effect was observed at a higher concentration (5 μM) compared to its GSK-3β IC50 (5 nM).
Another example of the NRF2 induction capacity of GSK-3β inhibitors is compound SB216763, which is a highly potent and selective GSK-3β inhibitor. SB216763 showed an interesting cytoprotective effect against the toxicity induced by doxorubicin in primary podocytes via the pathological activation of GSK-3β and oxidative stress [155]. SB216763 protected podocytes through NRF2 induction and phase II antioxidant response activation, and this response was directly associated to its GSK-3β inhibition capacity [155]. Finally, NRF2 induction via GSK-3β inhibition has been demonstrated for many other GSK-3β inhibitors such us YQ138 [156], obacunone [157], or corilagin [158]; however, in the latter cases, the NRF2 mechanism of action can also be related to a direct interaction with KEAP1 or other activation pathways due to the structural features and multi-target activity of the compounds.
3.2.2. KEAP1-Independent Regulation of NRF2 Stability
Besides KEAP1, there are other mechanisms that control the stability of NRF2. The most important ones are the β-transducin repeat containing protein (β-TrCP), connected to the PI3K/AKT pathway, and Hrd1 (Figure 11).
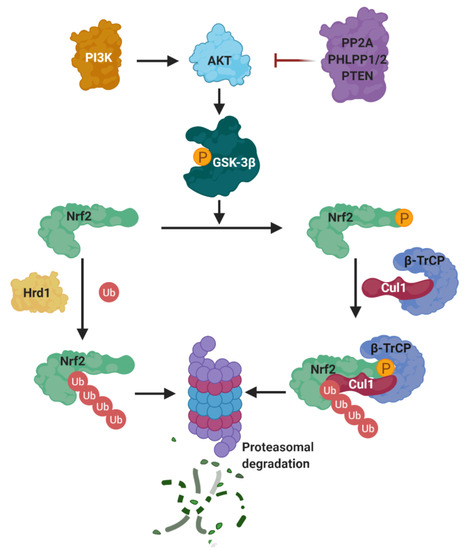
Figure 11. KEAP1-independent negative regulation of NRF2. The latter protein is tightly regulated by different systems, independently of its principal repressor KEAP1, to ensure the activation of the phase II response only under stress conditions. From left, the activation of different pathways might lead to PI3K activation that activates AKT by phosphorylation that in turn, from right, was inhibited by protein phosphatase 2A (PP2A), PHLPP1/2 (PH domain and leucine rich repeat protein phosphatases 1 and 2), or tumor suppressor phosphatase and tensin homolog (PTEN). After its activation, AKT is able to phosphorylate GSK-3β at Ser9 inhibiting its activity reducing the negative regulation of NRF2 by this kinase. GSK-3β is known to act as a negative NRF2 regulator by several mechanisms. NRF2 can be phosphorylated by this kinase at Ser335 and S338 to create a phosphodegron domain DSGIS recognized by β-TrCP that leads to Cul1/Rbx1 NRF2 ubiquination and proteasomal degradation. NRF2 is also regulated by a third ubiquitination-degradation system by interaction with Hrd1, at the Neh4 transactivation domain, which is a ubiquitin ligase that targets NRF2 for proteasomal degradation.
(a) PI3K/Akt. The PI3K/Akt pathway regulates metabolism, growth, proliferation, cell survival, and gene transcription. Akt is a Ser/Thr kinase activated by several signals that produce phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) catalyzed by phosphatidylinositol-3-kinase (PI3K) [159]. PIP3 serves as anchor for Akt and its activator kinase, PDK1, which activates Akt by phosphorylation at Thr308. Phosphorylated Akt is further activated by mTORC2 by phosphorylation at Ser473 [159]. A negative regulation of Akt is exerted by protein phosphatase 2A (PP2A), PHLPP1/2, and the tumor suppressor phosphatase and tensin homolog (PTEN) [159].
The PI3K/Akt pathway contributes to Nrf2 induction in a KEAP1-independent mode, as described by Cuadrado et al. [160]. By using the PI3K inhibitor LY294002, they observed a substantial inhibition of carnosol-mediated Nrf2 induction. Further evidence to support the regulation of Nrf2 induction by PI3K activation was obtained by using a biotinylated analogue of CDDO-Im, which is a potent Nrf2 inducer belonging to the cyanoenone triterpenoid family that activates the PI3K/Akt pathway because it reacts covalently with Cys124 inside the active site of PTEN [161], inhibiting its activity. Similarly, 4-hidroxynonenal (an Nrf2 inducer) modifies PTEN Cys71 [162]. The Cys71 and Cys124 residues of PTEN act as redox sensors and, when oxidized, they form a disulfide bridge inactivating the PIP3 3-phosphatase activity of PTEN and increasing the activity of Akt [163]. Therefore, PTEN is considered a negative repressor of Nrf2, acting as a redox sensor inside the cell; once it is inactivated, Nrf2 is activated via the PI3K/Akt pathway activation.
It was demonstrated that PI3K activation leads to Nrf2 induction [143][164] by increasing Akt activity that, in turn, inhibits the activity of GSK-3β by phosphorylation at Ser9 [165] and Ser21 [166]. Furthermore, GSK-3β can also be inhibited by PKC [167], P70S6K, and P90RSK [168], all of which are regulated by PI3K through PDK1 [169].
(b) β-TrCP. The PI3K/AKT pathway activates NRF2 via the inhibition of GSK-3β, which phosphorylates NRF2 [160], allowing its recognition by β-TrCP, which in turn marks NRF2 for ubiquitinylation and subsequent proteasomal degradation [170]. β-TrCP is an F-Box-containing protein of the WD40 subfamily that acts as substrate adaptor/receptor within the Skp1-Cul1-F-box (SCF) ubiquitin ligase SCFβ-TrCP [171] and participates in the ubiquination of IκB, β-catenin, CDC25, and APC [38], among others, besides NRF2. β-TrCP recognizes its substrates by direct binding to phosphorylated destruction motifs; for instance, it binds to phosphorylated NRF2 at its 343DSGIS347 and 382DSAPGS387 sequences [172]. Interestingly, this sequence mimics the GSK-3β target motifs, and thus many of the GSK-3β target proteins are known to be processed by β-TrCP for degradation [173].
By using selective GSK-3β inhibitors or siRNAs targeting GSK-3β, a stabilization and nuclear localization of NRF2 was initially observed. Similar results were obtained by using siRNAs toward β-TrCP, demonstrating the regulation of NRF2 by both proteins. The Neh6 domain of NRF2 presents two motifs, DSGIS and DSAPGS, that can be recognized by SCFβ-TrCP once they are phosphorylated [174]. Thus, the GSK-3β/β-TrCP system induces NRF2 ubiqination via β-TrCP/Cul1in1/Rbx1 E3 ligase complex after phosphorylation, being a KEAP1-independent mechanism activated by receptor signal transduction [175]. Considering this regulatory mechanism, a hypothesis of “dual modulation” by KEAP1 and β-TrCP was proposed by Cuadrado et al. [45]. Therefore, β-TrCP is a fine tune regulator of NRF2 for cell signaling, considering the high number of proteins regulated by Nr2 with a high impact on metabolic adaptation [46].
(c) DJ-1. In another connection to NDDs, a direct link between DJ-1, a protein involved in PD, and Nrf2 has been described. DJ-1 inactivation in PD is related to a higher susceptibility of neurons to oxidative stress [176]. Compelling evidence demonstrates that DJ-1 acts as a negative regulator of PTEN [177]. Therefore, DJ-1 downregulation reduces Nrf2 activity due to the increased activity of PTEN. Furthermore, DJ-1 presents several ROS-sensitive Cys residues that can act as oxidative stress sensors. Finally, it has been proposed that DJ-1 can act as a nitric oxide-transferring protein to PTEN Cys residues to inhibit its activity [178].
3.2.3. Miscellaneous ARE Transcription Regulators Connected to NRF2
(a) AHR. The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor of the bHLH/PAS family that mediates the effects of many xenobiotics (e.g., polyaromatic hydrocarbons, dioxins) and endogenous compounds [179]. It is widespread in the nervous system and, among others, regulates neuronal functions [179]. Miao et al. showed that the transcription of NRF2 is directly regulated by AHR [180], and in fact, they exhibit both ARE and XRE response elements in their promoter region. Moreover, they share several common antioxidant target genes [181].
(b) P53. The P53 protein, known as the guardian of the genome, is a transcription factor that is activated by DNA damage, regulating the pathways for cell-cycle arrest, DNA repair, senescence, and apoptosis. Indeed, it has been proposed that p53 may contribute to neuronal death processes common to all NDDs [182]. There are several connections between the NRF2 and p53 pathways [183]. First, the activation of p53 induces the expression of p21, which competes with KEAP1 for binding to NRF2 and prevents NRF2 degradation, rising its levels [184]. Moreover, NRF2 induces the expression of NQO1, which hinders the degradation of p53 by the 20S proteasome [185].
(c) Epigenetic regulation. As in the case of KEAP1, the transcription of NRF2 is subject to epigenetic regulation via methylation of the NRF2 promoter in CpG islands, H3 histone methylation, and H4 histone acetylation [186]. There is some evidence showing that DNA-methyltransferase inhibition during brain development increases susceptibility to oxidative DNA damage in the aged brain via the hypomethylation of promoters of AD-associated genes such as the β-amyloid precursor protein [187]. Interestingly, DNA demethylation by treatment with 5-azacytidine (31, Figure 12) was shown to upregulate NRF2 expression in an AD cellular model. [188] In the same context, it has been proven that one of the mechanisms of neuroprotection by sulforaphane (1, Figure 12) in mouse neuroblastoma cells is an upregulated NRF2 expression and promoted NRF2 nuclear translocation by decreasing the methylation levels of the NRF2 promoter gene [189].
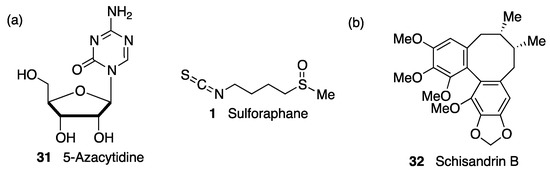
Figure 12. (a) Two compounds that exerted neuroprotection by epigenetically-promoted upregulation of NRF2 expression. (b) Schisandrin B exerted neuroprotection in a Parkinson’s disease (PD) cell model by blocking the suppression of NRF2 activity by the microRNA miR-34a.
NRF2 activity can also be regulated by microRNAs, which repress protein production by interacting with complementary seed sequences in the UTR of target mRNAs. Several miRs are involved in direct NRF2 regulation and are related with neurological disorders associated with oxidative stress [138]. Thus, miR-27a, miR-142-5p, miR-144, and miR-153 were identified as NRF2 suppressors by in silico analysis. Studies in neuronal SH-SY5Y cells showed that the overexpression of these miRs suppresses NRF2 activity by targeting its mRNA, leading to uncontrolled redox homeostasis [190]. Accordingly, schisandrin B (32, Figure 12), a dibenzocyclooctadiene lignan found in the traditional Chinese medicinal herb Schisandra chinensis, has shown neuroprotective effects in 6-OHDA PD models by acting as an inhibitor of miR-34a action [191]. Many miRs are involved in the pathological events after cerebral stroke, and among them, miR 93 has been found to be a negative regulator of Nfr2 expression that contributes significantly to deregulation of the redox balance in ischemia [192]. On the other hand, miR-424 upregulates NRF2, and its overexpression has an antioxidant effect in focal cerebral ischemia/reperfusion in mice that is suppressed by NRF2 knockdown and treatment with SOD inhibitors [193].
3.2.4. Miscellaneous Pathways Regulating the Post-Translational Phosphorylation of NRF2
Beyond the canonical pathway, the tight control of the phase II antioxidant response inside the cells encompasses a plethora of pathways that regulate NRF2 by different post-translational mechanisms [194][195]. Different positive and negative NRF2 regulation proteins have been described including the previously discussed GSK-3β [143], PI3K/Akt [160], MAPKs [196][197], β-TrCP [160], and PKC [198][199], among others. This fine-tuned control over NRF2 through non-canonical mechanisms opens up the opportunity to design drugs aimed at specific targets.
(a) MAPKs. These enzymes are serine/threonine kinases that modulate crucial biological processes including cell survival [196][200], apoptosis [198][201], and gene expression [197][200]. There are three main MAPK pathways composed by the ERKs: the c-jun N-terminal kinases (JNKs) and the p38 kinases directed to phosphorylation of Ser or Thr residues near to Pro residues [197]. NRF2 regulation by different MAPK pathways has been widely described by the use of selective inhibitors and transfection studies, which demonstrated that ERK signaling increases NRF2 activity [202] and P38 reduces NRF2 signaling [203]. Sun et al. demonstrated that a number of phosphorylation sites at NRF2, including Ser215, Ser408, Ser558, Thr559 and S577, are post-transcriptionally modified by the members of this family of kinases [204].
(b) P38. The P38 MAPK family comprises several isoforms (P38α, β, γ and δ) with specific distribution and substrates. Once activated by phosphorylation, they in turn phosphorylate different proteins and transcription factors to regulate processes including inflammation, cell division, and differentiation or apoptosis [205]. P38 kinases are mainly activated by oxidative stress [206], which is a pathological hallmark of NDDs. P38 has been related to AD, since it is activated by the Aβ peptide and hyperphosphorylated tau through oxidative stress [207]. In this line, the P38 pathway was described to be overactive in AD brains at initial stages [208]. The general consensus points to a negative regulation of NRF2 by P38 MAPK as shown by Keum et al., who described the active p38 isoforms that are able to phosphorylate the NRF2 protein to reduce its nuclear accumulation [209]. P38 MAPK phosphorylates NRF2 at three Ser residues (Ser215, Ser408, and Ser577), improving its interaction with KEAP1 and thus promoting its degradation [6]. Conversely, p38 activation activated the NRF2–ARE pathway [210][211] after the administration of several xenobiotics, which is an observation that was correlated with the inhibitory activity of P38 on GSK-3β [209]. Most probably, the activity of P38 on NRF2 depends of small modifications of the homeostatic equilibrium, and the final response depends on the stimuli, timing, and cellular type.
(c) JNK. The c-jun-NH2-terminal kinase (JNK) pathway is activated mainly by stress stimuli and is an important member of the MAPK signaling pathways [212], playing important roles in development, cell growth, inflammation, and apoptosis [213]. JNKs are a family of threonine protein kinases that includes three different isoforms (JNK1, JNK2, and JNK3), the latter of which is specifically expressed in the central nervous system [214] and is considered a key target for NDDs. JNK3 has been found to be activated in AD [215], PD [216], ALS, and stroke [217], among other NDDs. Sustained JNK activation has been widely related to pathological conditions promoting cell damage and death and has also been correlated with decreased NRF2 signaling. Recently, a direct crosstalk between JNK and NRF2 has been revealed in the context of protection against liver injury induced by acetaminophen, which is closely associated to JNK activation [218]. Active JNK (p-JNK) increased NRF2 turnover by direct interaction with the Neh1 domain of NRF2. This interaction allows the phosphorylation of Ser residues at Neh6 degron domain of NRF2. Thus, it was demonstrated that p-JNK phosphorylates Ser335 at DSGIS motif of NRF2, codifying its degradation through a KEAP1-independent mechanism [219].
(d) ERKs. The extracellular signal-regulated kinases (ERKs) are a major MAPK subfamily involved in a large number of biological processes. ERK activation has been highly related to cytoprotection, specifically to neuroprotection, linking this kinase pathway to potential targets against NDDs [220]. NRF2 regulation by the ERK pathway has been reported in many different cellular types, and it has been implicated in the mechanism of action of several NRF2 inducers. tBHQ, a known NRF2 inducer [102], increases NRF2 stability inducing the activation of the phase II response in a ERK1/2-dependent mechanism, since its NRF2 induction capacity was abolished in the presence of ERK inhibitors [221]. ERK activation was demonstrated to be necessary for NRF2–ARE signaling in response to several other NRF2 inducers such as pyrrolidine dithiocarbamate [222], caffeic acid [223], sulfuretin [224], gallic acid [224], luteolin [225], and butylated hydroxyanisole [226] in HepG2 cells; lico A in RAW 264.7 cells (26576227), or phenylethanoid glycoside in PC12 cells. Some reports suggest a potential direct crosstalk between NRF2 and ERK1/2 by which activated ERK1/2 might be able to directly phosphorylate NRF2 at Ser40, codifying its nuclear stabilization to increase NRF2–ARE pathway activation [227]. This hypothesis is supported by an increase of p-Ser40-NRF2 protein levels measured by Western blot. However, a direct interaction of ERK1/2 and NRF2 has not been fully proven, and other pathways might be contributing such as PKC, JNK, or Akt [227].
(e) PKC. This protein is part of a family of serine/threonine kinases related to many cellular signaling responses including apoptosis, survival, and differentiation. Although PKC activation is traditionally considered receptor-dependent, there is also a ROS-dependent activation mechanism [228]. The discovery that phorbol esters (PKC activators) induce the expression of NRF2-related proteins led to hypothesize the regulation of NRF2-phase II response by PKC [198]. It was proved that, together with other kinases [199], PKC phosphorylates Ser40 at the NRF2Neh2 domain, disrupting the KEAP1–NRF2 interaction [229]. Ser40 phosphorylation is necessary for its nuclear translocation, but it does not facilitate its nuclear accumulation [230]. A second NRF2 regulation mechanism by PKC is related to the capacity of several PKC isoforms (α, β1, γ > β2; not ε) to phosphorylate GSK-3β at inactivation sites [231]. Focusing on NDDs, PKC protein levels and activity was found to be decreased in AD brains [232]. Considering the positive modulation of NRF2 by PKC and other potentially beneficial properties, several PKC activators have been proposed as potential drugs for AD treatment [233].
(f) CK2. Casein kinase II (CK2) is a Ser/Thr kinase that acts on more than 300 substrates, participating in the regulation of many different processes including signal transduction, gene transcription, replication processes, and survival pathways, among others. Among the processes modulated by CK2, it regulates several key regulators against cell stress [234]. CK2 is also related to NRF2 modulation, since its sequence presents at least 13 potential phosphorylation positions targeted by CK2 [235]. By using deletional analyses of NRF2, Neh4 and Neh5 domains were identified as CK2 target regions where this kinase is able to phosphorylate several residues, inducing NRF2 nuclear accumulation and pathway activation [235].
(g) Fyn kinase. Fyn kinase, a member of the Src family, is closely related to the negative regulation of NRF2. Fyn kinase directly phosphorylates NRF2 at Tyr568 to codify its nuclear export [236]. Tyr568 phosphorylation facilitates NRF2 recognition by exportin1, which is the protein related to NRF2 nuclear elimination [236].
References
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (NRF2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl Acad. Sci. USA 1994, 91, 9926–9930.
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An NRF2/small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322.
- Hayes, J.D.; Dinkova-Kostova, A.T. The NRF2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. KEAP1 represses nuclear activation of antioxidant responsive elements by NRF2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86.
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The KEAP1-BTB protein is an adaptor that bridges NRF2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-KEAP1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharm. Rev. 2018, 70, 348–383.
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of NRF2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590.
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948.
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by NRF2. Antioxid. Redox Signal. 2018, 29, 1727–1745.
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 2005, 25, 8044–8051.
- Fuse, Y.; Kobayashi, M. Conservation of the KEAP1-NRF2 system: An evolutionary journey through stressful space and time. Molecules 2017, 22, 436.
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. KEAP1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900.
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of NRF2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906.
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of NRF2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868.
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the NRF2 transcription factor in a KEAP1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133.
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108.
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317.
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded NRF2 activation on phase-I and-II drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 2012, 7, e39006.
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor NRF2. J. Biol. Chem. 1999, 274, 33627–33636.
- Pajares, M.; Jiménez-Moreno, N.; García-Yague, A.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916.
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of NRF2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188.
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. NRF2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624.
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L.J. Expression of Nrf2 in neurodegenerative diseases. Neuropathol. Exp. Neurol. 2007, 66, 75–85.
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. NRF2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938.
- Sarlette, A.; Kramp, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062.
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rabano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91.
- Cuadrado, A.; Moreno-Murciano, P.; Pedraza-Chaverri, J. The transcription factor NRF2 as a new therapeutic target in Parkinson’s disease. Expert Opin. Targets 2009, 13, 319–329.
- Dumont, M.; Wille, E.; Calingasan, N.Y.; Tampellini, D.; Williams, C.; Gouras, G.K.; Liby, K.; Sporn, M.; Nathan, C.; Beal, M.F.; et al. Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2009, 109, 502–512.
- Kaidery, N.A.; Banerjee, R.; Yang, L.; Smirnova, N.A.; Hushpulian, D.M.; Liby, K.T.; Williams, C.R.; Yamamoto, M.; Kensler, T.W.; Ratan, R.R.; et al. Targeting NRF2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal. 2013, 18, 139–157.
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of NRF2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96.
- Kumar, H.; Koppula, S.; Kim, I.S.; More, S.V.; Kim, B.W.; Choi, D.K. Nuclear factor erythroid 2-related factor 2 signaling in Parkinson disease: A promising multi therapeutic target against oxidative stress, neuroinflammation and cell death. CNS Neurol. Disord. Drug Targets 2012, 11, 1015–1029.
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of NRF2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47.
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox. Biol. 2017, 11, 543–553.
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The NRF2 cell defence pathway: KEAP1-dependent and independent mechanisms of regulation. Biochem. Pharm. 2013, 85, 705–717.
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239.
- Snyder, G.H.; Cennerazzo, M.J.; Karalis, A.J.; Field, D. Electrostatic influence of local cysteine environments on disulfide exchange kinetics. Biochemistry 1981, 20, 6509–6519.
- Holland, R.; Fishbein, J.C. Chemistry of the cysteine sensors in Kelch-like ECH-associated protein 1. Antioxid. Redox Signal. 2010, 13, 1749–1761.
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of KEAP1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. USA 2002, 99, 11908–11913.
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in KEAP1 are required for KEAP1-dependent ubiquitination of NRF2 and for stabilization of NRF2 by chemo-preventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151.
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. KEAP1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc and alkenals. Proc. Natl. Acad. USA 2010, 107, 18838–18843.
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of NRF2 activation. Pharm. Res. 2018, 134, 92–99.
- Eggler, A.L.; Gay, K.A.; Mesecar, A.D. Molecular mechanisms of natural products in chemoprevention: Induction of cytoprotective enzymes by NRF2. Mol. Nutr. Food Res. 2008, 52, 84–94.
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the KEAP1 sensor modified by inducers. Proc. Natl. Acad. USA 2004, 101, 2040–2045.
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the NRF2-KEAP1 complex. J. Biol. Chem. 2006, 281, 24756–24768.
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y.; You, Q.D. The KEAP1-NRF2-ARE pathway as a potential preventive and therapeutic target: An update. Med. Res. Rev. 2016, 36, 924–963.
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the NRF2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic. Biol. Med. 2016, 99, 544–556.
- Hur, W.; Gray, N.S. Small molecule modulators of antioxidant response pathway. Curr. Opin. Chem. Biol. 2011, 15, 162–173.
- Satoh, T.; McKercher, S.R.; Lipton, S.A. NRF2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657.
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent modification at Cys151 dissociates the electrophile sensor KEAP1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710.
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of three major cysteine sensors of KEAP1 in stress response. Mol. Cell. Biol. 2015, 36, 271–284.
- Zhou, Y.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent advances of natural polyphenols activators for KEAP1-NRF2 signaling pathway. Chem. Biodivers. 2019, 16, e1900400.
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821.
- Mori, T.; Ishii, T.; Akagawa, M.; Nakamura, Y.; Nakayama, T. Covalent binding of tea catechins to protein thiols: The relationship between stability and electrophilic reactivity. Biosci. Biotechnol. Biochem. 2010, 74, 2451–2456.
- Yang, G.Z.; Wang, Z.J.; Bai, F.; Qin, X.J.; Cao, J.; Lv, J.Y.; Zhang, M.S. Epigallocatechin-3-gallate protects HUVECs from PM2.5-induced oxidative stress injury by activating critical antioxidant pathways. Molecules 2015, 20, 6626–6639.
- Ma, L.; Cao, T.T.; Kandpal, G.; Warren, L.; Fred Hess, J.; Seabrook, G.R.; Ray, W.J. Genome-wide microarray analysis of the differential neuroprotective effects of antioxidants in neuroblastoma cells overexpressing the familial Parkinson’s disease alpha-synuclein A53T mutation. Neurochem. Res. 2010, 35, 130–142.
- Itoh, T.; Tabuchi, M.; Mizuguchi, N.; Imano, M.; Tsubaki, M.; Nishida, S.; Hashimoto, S.; Matsuo, K.; Nakayama, T.; Ito, A.; et al. Neuroprotective effect of (-)-epigallocatechin-3-gallate in rats when administered pre- or post-traumatic brain injury. J. Neural. Transm. 2013, 120, 767–783.
- Xu, Q.; Kanthasamy, A.G.; Reddy, M.B. Epigallocatechin gallate protects against TNFalpha-or H2O2-induced apoptosis by modulating iron related proteins in a cell culture model. Int. J. Vitam. Nutr. Res. 2018, 88, 158–165.
- Dinkova-Kostova, A.T.; Talalay, P.; Sharkey, J.; Zhang, Y.; Holtzclaw, W.D.; Wang, X.J.; David, E.; Schiavoni, K.H.; Finlayson, S.; Mierke, D.F.; et al. An exceptionally potent inducer of cytoprotective enzymes: Elucidation of the structural features that determine inducer potency and reactivity with KEAP1. J. Biol. Chem. 2010, 285, 33747–33755.
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate NRF2 through inhibition of ubiquitination activity of KEAP1. Mol Cell Biol. 2006, 26, 221–229.
- Sen, C.K.; Sashwati, R.; Packer, L. Fas mediated apoptosis of human Jurkat T-cells: Intracellular events and potentiation by redox-active alpha-lipoic acid. Cell Death Differ. 1999, 6, 481–491.
- Chaudhary, P.; Marracci, G.; Galipeau, D.; Pocius, E.; Morris, B.; Bourdette, D. Lipoic acid reduces inflammation in a mouse focal cortical experimental autoimmune encephalomyelitis model. J Neuroimmunol. 2015, 289, 68–74.
- Zhao, H.; Zhao, X.; Liu, L.; Zhang, H.; Xuan, M.; Guo, Z.; Wang, H.; Liu, C. Neurochemical effects of the R form of alpha-lipoic acid and its neuroprotective mechanism in cellular models of Parkinson’s disease. Int. J. Biochem. Cell. Biol. 2017, 87, 86–94.
- Agarwal, N.B.; Jain, S.; Agarwal, N.K.; Mediratta, P.K.; Sharma, K.K. Modulation of pentylenetetrazole-induced kindling and oxidative stress by curcumin in mice. Phytomedicine 2011, 18, 756–759.
- Dong, W.; Yang, B.; Wang, L.; Li, B.; Guo, X.; Zhang, M.; Jiang, Z.; Fu, J.; Pi, J.; Guan, D.; et al. Curcumin plays neuroprotective roles against traumatic brain injury partly via NRF2 signaling. Toxicol. Appl. Pharm. 2018, 346, 28–36.
- Rao, M.N.A. Nitric oxide scavenging by curcuminoids. J. Pharm. Pharm. 1997, 49, 105–107.
- Ren, L.; Zhan, P.; Wang, Q.; Wang, C.; Liu, Y.; Yu, Z.; Zhang, S. Curcumin upregulates the NRF2 system by repressing inflammatory signaling-mediated KEAP1 expression in insulin-resistant conditions. Biochem. Biophys. Res. Commun. 2019, 514, 691–698.
- Wang, B.F.; Cui, Z.W.; Zhong, Z.H.; Sun, Y.H.; Sun, Q.F.; Yang, G.Y.; Bian, L.G. Curcumin attenuates brain edema in mice with intracerebral hemorrhage through inhibition of AQP4 and AQP9 expression. Acta Pharm. Sin. 2015, 36, 939–948.
- Zhang, L.; Wu, C.; Zhao, S.; Yuan, D.; Lian, G.; Wang, X.; Wang, L.; Yang, J. Demethoxycurcumin, a natural derivative of curcumin attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia induced by lipopolysaccharide. Int. Immunopharmacol. 2010, 10, 331–338.
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1091–1100.
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the KEAP1-NRF2 system. Free Radic. Biol. Med. 2012, 53, 817–827.
- Wang, W.; Wu, Y.; Zhang, G.; Fang, H.; Wang, H.; Zang, H.; Xie, T.; Wang, W. Activation of NRF2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 2014, 1544, 54–61.
- Duran, C.G.; Burbank, A.J.; Mills, K.H.; Duckworth, H.R.; Aleman, M.M.; Kesic, M.J.; Peden, D.B.; Pan, Y.; Zhou, H.; Hernandez, M.L. A proof-of-concept clinical study examining the NRF2 activator sulforaphane against neutrophilic airway inflammation. Respir. Res. 2016, 17, 89.
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and other nutrigenomic NRF2 activators: Can the clinician’s expectation be matched by the reality? Oxid. Med. Cell. Longev. 2016, 2016, 7857186.
- Park, H.M.; Kim, J.A.; Kwak, M.K. Protection against amyloid beta cytotoxicity by sulforaphane: Role of the proteasome. Arch. Pharmacal Res. 2009, 32, 109–115.
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y.S. Amelioration of Alzheimer’s disease by neuropro-tective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12.
- Han, J.M.; Lee, Y.J.; Lee, S.Y.; Kim, E.M.; Moon, Y.; Kim, H.W.; Hwang, O. Protective effect of sulfo-raphane against dopaminergic cell death. Pharm. L Exp. 2007, 321, 249–256.
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. NRF2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496.
- Buendia, I.; Navarro, E.; Michalska, P.; Gameiro, I.; Egea, J.; Abril, S.; López, A.; González-Lafuente, L.; López, M.G.; León, R. New melatonin-cinnamate hybrids as multi-target drugs for neurodegenerative diseases: NRF2-induction, antioxidant effect and neuroprotection. Future Med. Chem. 2015, 7, 1961–1969.
- Sun, H.; Zhu, J.; Lin, H.; Gu, K.; Feng, F. Recent progress in the development of small molecule NRF2 modulators: A patent review (2012–2016). Expert Opin. Pat. 2017, 27, 763–785.
- Zhang, D.D. Bardoxolone brings NRF2-based therapies to light. Antioxid. Redox Signal. 2013, 19, 517–518.
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26.
- Freeman, B.A.; O’Donnell, V.B.; Schopfer, F.J. The discovery of nitro fatty acids as products of metabolic and inflammatory reactions and mediators of adaptive cell signaling. Nitric Oxide 2018, 77, 106–111.
- Deen, A.J.; Sihvola, V.; Härkönen, J.; Patinen, T.; Adinolfi, S.; Levonen, A.L. Regulation of stress signaling pathways by nitro-fatty acids. Nitric Oxide 2018, 78, 170–175.
- Baker, L.M.; Baker, P.R.; Golin-Bisello, F.; Schopfer, F.J.; Fink, M.; Woodcock, S.R.; Branchaud, B.P.; Radi, R.; Freeman, B.A. Nitro- fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J. Biol. Chem. 2007, 282, 31085–31093.
- Schopfer, F.J.; Vitturi, D.A.; Jorkasky, D.K.; Freeman, B.A. Nitro-fatty acids: New drug candidates for chronic inflammatory and fibrotic diseases. Nitric Oxide 2018, 79, 31–37.
- Hoxtermann, S.; Nuchel, C.; Altmeyer, P. Fumaric acid esters suppress peripheral CD4-and CD8-positive lymphocytes in psoriasis. Dermatology 1998, 196, 223–230.
- Deeks, E.D. Dimethyl fumarate: A review in relapsing-remitting MS. Drugs 2016, 76, 243–254.
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952.
- Muller, S.; Behnen, M.; Bieber, K.; Moller, S.; Hellberg, L.; Witte, M.; Hansel, M.; Zillikens, D.; Solbach, W.; Laskay, T.; et al. Dimethyl fumarate impairs neutrophil functions. J. Invest. Dermatol. 2016, 136, 117–126.
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myslinska, D.; Grembecka, B.; Rucinski, J.; Pierzynowska, K.; Wrona, D. Age-dependent effects of dimethyl fumarate on cognitive and neuropathological features in the streptozotocin-induced rat model of Alzheimer’s disease. Brain Res. 2018, 1686, 19–33.
- Kunze, R.; Urrutia, A.; Hoffmann, A.; Liu, H.; Helluy, X.; Pham, M.; Reischl, S.; Korff, T.; Marti, H.H. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp. Neurol. 2015, 266, 99–111.
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 61–77.
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 2018, 22, 1081–1094.
- Bahn, G.; Jo, D.G. Therapeutic approaches to Alzheimer’s disease through modulation of NRF2. Neuromolecular Med. 2019, 21, 1–11.
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the NRF2 antioxidant pathway. Brain 2011, 134, 678–692.
- Zeidan, T.A.; Duncan, S.; Hencken, C.P.; Wynn, T.A.; Sanrame, C.N. Prodrugs of Fumarates and Their Use in Treating Various Diseases. US8669281B1, 3 November 2014.
- Zarling, J.A.; Brunt, V.E.; Vallerga, A.K.; Li, W.; Tao, A.; Zarling, D.A.; Minson, C.T. Nitroxide pharmaceutical development for age-related degeneration and disease. Front. Genet. 2015, 6, 00325.
- Cuzzocrea, S.; Pisano, B.; Dugo, L.; Ianaro, A.; Patel, N.S.; Caputi, A.P.; Thiemermann, C. Tempol reduces the activation of nuclear factor-kappaB in acute inflammation. Free. Radic. Res. 2004, 38, 813–819.
- Greenwald, B.Y.M.; Anzi, S.; Ben Sasson, S.; Bianco-Peled, H.; Kohen, R. Can nitroxides evoke the KEAP1-Nrf2-ARE pathway in skin? Free Rad. Med. Biol. 2014, 77, 258–279.
- Hirotsu, Y.; Katsuoka, F.; Itoh, K.; Yamamoto, M. NRF2 degron- fused reporter system: A new tool for specific evaluation of NRF2 inducers. Genes Cells 2011, 16, 406–415.
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor NRF2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie 2010, 92, 245–253.
- Sukumari-Ramesh, S.; Alleyne, C.H., Jr. Post-injury administration of tert-butylhydroquinone attenuates acute neurological injury after intracerebral hemorrhage in mice. J. Mol. Neurosci. 2016, 58, 525–531.
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel NRF2-inducer prevents mitochondrial defects and oxidative stress in Friedreich’s ataxia models. Front. Cell. Neurosci. 2018, 12, 188.
- Jiang, Z.Y.; Lu, M.C.; You, Q.D. Discovery and development of Kelch-like ECH-associated protein 1. Nuclear factor erythroid 2-related factor 2 (KEAP1: NRF2) protein-protein interaction inhibitors: Achievements, challenges, and future directions. J. Med. Chem. 2016, 59, 10837–10858.
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244.
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the KEAP1: NRF2 interface provides mechanistic insight into NRF2 signaling. EMBO J. 2006, 25, 3605–3617.
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-κB signaling by targeting IKKβ. Mol. Cell 2009, 36, 131–140.
- Zhang, Y.; Shi, Z.; Zhou, Y.; Xiao, Q.; Wang, H.; Peng, Y. Emerging substrate proteins of Kelch-like ECH associated protein 1 (KEAP1) and potential challenges for the development of small-molecule inhibitors of the KEAP1-nuclear factor erythroid 2-related factor 2 (NRF2) protein-protein interaction. J. Med. Chem. 2020, 63.
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, E.; Park, B.K. Physical and functional interaction of sequestosome 1 with KEAP1 regulates the KEAP1-NRF2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788.
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the KEAP1-NRF2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631.
- Lin, X.; Li, S.; Zhao, Y.; Ma, X.; Zhang, K.; He, X.; Wang, Z. Interaction domains of p62: A bridge between p62 and selective autophagy. DNA Cell Biol. 2013, 32, 220–227.
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the KEAP1-NRF2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61.
- Padmanabhan, B.; Nakamura, Y.; Yokoyama, S. Structural analysis of the complex of KEAP1 with a prothymosin α peptide. Acta Cryst. F 2008, 64, 233–238.
- Ueda, H.; Halder, S.K.; Matsunaga, H.; Sasaki, K.; Maeda, S. Neuroprotective impact of prothymosin alpha-derived hexapeptide against retinal ischemia-reperfusion. Neuroscience 2016, 318, 206–218.
- Karapetian, R.N.; Evstafieva, A.G.; Abaeva, I.S.; Chichkova, N.V.; Filonov, G.S.; Rubtsov, Y.P.; Sukhacheva, E.A.; Melnikov, S.V.; Schneider, U.; Wanker, E.E.; et al. Nuclear oncoprotein prothymosin α is a partner of KEAP1: Implications for expression of oxidative stress-protecting genes. Mol. Cell. Biol. 2005, 25, 1089–1099.
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa B kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819.
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-kappa B signaling by KEAP1 regulation of IKKβ activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654.
- Jiang, Z.Y.; Chu, H.X.; Xi, M.Y.; Yang, T.T.; Jia, J.M.; Huang, J.J.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Insight into the intermolecular recognition mechanism between KEAP1 and IKKβ combining homology modelling, protein-protein docking, molecular dynamics simulations and virtual alanine mutation. PLoS ONE 2013, 8, e75076.
- Wells, G. Peptide and small molecule inhibitors of the Keap1-Nrf2 protein-protein interaction. Biochem. Soc. Trans. 2015, 43, 674–679.
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of KEAP1-NRF2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043.
- Ontoria, J.M.; Biancofiore, I.; Fezzardi, P.; Ferrigno, F.; Torrente, E.; Colarusso, S.; Bianchi, E.; Andreini, M.; Patsilinakos, A.; Kempf, G.; et al. Combined peptide and small-molecule approach towards non-acidic THIQ inhibitors of the KEAP1/NRF2 interaction. ACS Med. Chem. Lett. 2020.
- Jain, A.D.; Potteti, H.; Richardson, B.G.; Kingsley, L.; Luciano, J.P.; Ryuzoji, A.F.; Lee, H.; Krunic, A.; Mesecar, A.D.; Reddy, S.P.; et al. Probing the structural requirements of non-electrophilic naphthalene-based NRF2 activators. Eur. J. Med. Chem. 2015, 103, 252–268.
- Richardson, B.G.; Jain, A.D.; Potteti, H.R.; Lazzara, P.R.; David, B.P.; Tamatam, C.R.; Choma, E.; Skowron, K.; Dye, K.; Siddiqui, Z.; et al. Replacement of a naphthalene scaffold in Kelch-like ECHAssociated protein 1 (KEAP1)/nuclear factor (erythroid-derived 2)-like 2 (NRF2) inhibitors. J. Med. Chem. 2018, 61, 8029–8047.
- Lu, M.C.; Tan, S.J.; Ji, J.A.; Chen, Z.Y.; Yuan, Z.W.; You, Q.D.; Jiang, Z.Y. Polar recognition group study of KEAP1-NRF2 protein-protein interaction inhibitors. ACS Med. Chem. Lett. 2016, 7, 835–840.
- Shimozono, R.; Asaoka, Y.; Yoshizawa, Y.; Aoki, T.; Noda, H.; Yamada, M.; Kaino, M.; Mochizuki, H. NRF2 activators attenuate the progression of nonalcoholic steatohepatitis-related fibrosis in a dietary rat model. Mol. Pharm. 2013, 84, 62–70.
- Zhuang, C.L.; Narayanapillai, S.; Zhang, W.N.; Sham, Y.Y.; Xing, C.G. Rapid identification of KEAP1-NRF2 small-molecule inhibitors through structure based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126.
- Kim, S.; Viswanath, A.N.I.; Park, J.H.; Lee, H.E.; Park, A.Y.; Choi, J.W.; Kim, H.J.; Londhe, A.M.; Jang, B.K.; Lee, J.; et al. NRF2 activator via interference of NRF2-KEAP1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 2020, 167, 107989.
- Davies, T.G.; Wixted, W.E.; Coyle, J.E.; Griffiths-Jones, C.; Hearn, K.; McMenamin, R.; Norton, D.; Rich, S.J.; Richardson, C.; Saxty, G.; et al. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: Nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 2016, 59, 3991–4006.
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, synthesis, and evaluation of triazole derivatives that induce NRF2 dependent gene products and inhibit the KEAP1-NRF2. J. Med. Chem. 2015, 58, 7186–7194.
- Kazantsev, A.G.; Thompson, L.M.; Abagyan, R.; Casale, M. Small Molecule Activators of NRF2 Pathway. WO2014197818 A2, 3 November 2014.
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2-and NRF2-targeting thiazole-containing compound with therapeutic activity in Huntington’s disease models. Cell. Chem. Biol. 2016, 23, 849–861.
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046.
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49.
- Yildirim, F.; Gertz, K.; Kronenberg, G.; Harms, C.; Fink, K.B.; Meisel, A.; Endres, M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp. Neurol. 2008, 210, 531–542.
- Faraco, G.; Pancani, T.; Formentini, L.; Mascagni, P.; Fossati, G.; Leoni, F.; Moroni, F.; Chiarugi, A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharm. 2006, 70, 1876–1884.
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor NRF2 and protects against cerebral ischemic damage. Free Radic Biol Med. 2012, 52, 928–936.
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. NRF2 pathway in age-related neurological disorders: Insights into microRNAs. Cell. Physiol. Biochem. 2018, 47, 1951–1976.
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates NRF2 pathway by targeting KEAP1 expression. Free Radic. Biol. Med. 2015, 89, 548–556.
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057.
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118664.
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62.
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851.
- Jain, A.K.; Jaiswal, A.K. GSK-3β acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510.
- Velichkova, M.; Hasson, T. KEAP1 regulates the oxidation-sensitive shuttling of NRF2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol. Cell. Biol. 2005, 25, 4501–4513.
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of NRF2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499.
- Shaw, M.; Cohen, P. Role of protein kinase B and the MAP kinase cascade in mediating the EGF-dependent inhibition of glycogen synthase kinase 3 in Swiss 3T3 cells. Febs. Lett. 1999, 461, 120–124.
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704.
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK-3β inactivation. Science 2008, 320, 667–670.
- Eldar-Finkelman, H.; Martínez, A. GSK-3 Inhibitors: Preclinical and clinical focus on CNS. Front. Mol. Neurosci. 2011, 4, 32.
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium promotes longevity through GSK-3/NRF2-dependent hormesis. Cell Rep. 2016, 15, 638–650.
- Hu, B.; Wu, Y.; Liu, J.; Shen, X.; Tong, F.; Xu, G.; Shen, R. GSK-3β inhibitor induces expression ofNRF2/TrxR2 signaling pathway to protect against renal ischemia/reperfusion injury in diabetic rats. Kidney Blood Press. Res. 2016, 41, 937–946.
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88.
- Armagan, G.; Sevgili, E.; Gurkan, F.T.; Kose, F.A.; Bilgic, T.; Dagci, T.; Saso, L. Regulation of the NRF2 pathway by glycogen synthase kinase-3β in MPP(+)-induced cell damage. Molecules 2019, 24, 1377.
- Zhou, S.; Wang, P.; Qiao, Y.; Ge, Y.; Wang, Y.; Quan, S.; Yao, R.; Zhuang, S.; Wang, L.J.; Du, Y.; et al. Genetic and pharmacologic targeting of glycogen synthase kinase 3β reinforces the NRF2 antioxidant defense against podocytopathy. J. Am. Soc. Nephrol. 2016, 27, 2289–2308.
- Pang, T.; Wang, Y.J.; Gao, Y.X.; Xu, Y.; Li, Q.; Zhou, Y.B.; Xu, L.; Huang, Z.J.; Liao, H.; Zhang, L.Y.; et al. A novel GSK-3β inhibitor YQ138 prevents neuronal injury induced by glutamate and brain ischemia through activation of the NRF2 signaling pathway. Acta Pharm. Sin. 2016, 37, 741–752.
- Zhou, J.; Wang, T.; Wang, H.; Jiang, Y.; Peng, S. Obacunone attenuates high glucose-induced oxidative damage in NRK-52E cells by inhibiting the activity of GSK-3β. Biochem. Biophys. Res. Commun. 2019, 513, 226–233.
- Lv, H.; Hong, L.; Tian, Y.; Yin, C.; Zhu, C.; Feng, H. Corilagin alleviates acetaminophen-induced hepatotoxicity via enhancing the AMPK/GSK-3β-NRF2 signaling pathway. Cell. Commun. Signal. 2019, 17, 2.
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383.
- Martín, D.; Rojo, A.I.; Salinas, M.; Díaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929.
- Pitha-Rowe, I.; Liby, K.; Royce, D.; Sporn, M. Synthetic triterpenoids attenuate cytotoxic retinal injury: Cross-talk between Nrf2 and PI3K/AKT signaling through inhibition of the lipid phosphatase PTEN. Invest. Ophthalmol. Vis. Sci. 2009, 50, 5339–5347.
- Shearn, C.T.; Smathers, R.L.; Backos, D.S.; Reigan, P.; Orlicky, D.J.; Petersen, D.R. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic. Biol. Med. 2013, 65, 680–692.
- Worby, C.A.; Dixon, J.E. Pten. Annu. Rev. Biochem. 2014, 83, 641–669.
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678.
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19.
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789.
- Goode, N.; Hughes, K.; Woodgett, J.R.; Parker, P.J. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J. Biol. Chem. 1992, 267, 16878–16882.
- Cohen, P.; Frame, S. The renaissance of GSK-3. Nat. Rev. Mol. Cell. Biol. 2001, 2, 769–776.
- Bayascas, J.R. PDK1: The major transducer of PI 3-kinase actions. Curr. Top. Microbiol. Immunol. 2010, 346, 9–29.
- Hayes, J.D.; Ebisine, K.; Sharma, R.S.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Regulation of the CNC-bZIP transcription factor Nrf2 by KEAP1 and the axis between GSK-3 and β-TrCP. Curr. Opin. Toxicol. 2016, 1, 92–103.
- Fuchs, S.Y.; Spiegelman, V.S.; Suresh Kumar, K.G. The many faces of β-TrCP E3 ubiquitin ligases: Reflections in the magic mirror of cancer. Oncogene 2004, 23, 2028–2036.
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781.
- Robertson, H.; Hayes, J.D.; Sutherland, C. A partnership with the proteasome; The destructive nature of GSK-3. Biochem. Pharm. 2018, 147, 77–92.
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of NRF2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567.
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 1999, 13, 270–283.
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096.
- Kim, R.H.; Peters, M.; Jang, Y.; Shi, W.; Pintilie, M.; Fletcher, G.C.; DeLuca, C.; Liepa, J.; Zhou, L.; Snow, B.; et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 2005, 7, 263–273.
- Choi, M.S.; Nakamura, T.; Cho, S.J.; Han, X.; Holland, E.A.; Qu, J.; Petsko, G.A.; Yates, J.R.; Liddington, R.C.; Lipton, S.A. Transnitrosylation from DJ-1 to PTEN attenuates neuronal cell death in Parkinson’s disease models. J. Neurosci. 2014, 34, 15123–15131.
- Juricek, L.; Coumoul, X. The aryl hydrocarbon receptor and the nervous system. Int. J. Mol. Sci. 2018, 19, 2504.
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348.
- Dietrich, C. Antioxidants functions of the aryl hydrocarbon receptor. Stem. Cells Int. 2016, 2016, 10.
- Szybińska, A.; Leśniak, W. P53 dysfunction in neurodegenerative diseases-The cause or effect of pathological changes? Aging Dis. 2017, 8, 506–518.
- Hiemstra, S.; Niemeijer, M.; Koedoot, E.; Wink, S.; Klip, J.E.; Vlasveld, M.; de Zeeuw, E.; van Os, B.; White, W.; van de Water, B. Comprehensive landscape of NRF2 and p53 pathway activation dynamics by oxidative stress and DNA damage. Chem. Res. Toxicol. 2017, 30, 923–933.
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between NRF2 and p21(Cip1/WAF1) upregulates the NRF2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673.
- Chen, W.; Jiang, T.; Wang, H.; Tao, S.; Lau, A.; Fang, D.; Zhang, D.D. Does NRF2 contribute to p53-mediated control of cell survival and death? Antioxid. Redox Sign. 2012, 17, 1670–1675.
- Guo, Y.; Yu, S.; Zhang, C.; Kong, A.-N.T. Epigenetic regulation of KEAP1-NRF2 signaling. Free Radical Biol. Med. 2015, 88, 337–349.
- Zawia, N.; Lahiri, D.K.; Cardozo-Peláez, F. Epigenetics, oxidative stress, and Alzheimer’s disease. Free Radical Biol. Med. 2009, 46, 1241–1249.
- Cao, H.; Wang, L.; Chen, B.; Zheng, P.; He, Y.; Ding, Y.; Deng, Y.; Lu, X.; Guo, X.; Zhang, Y.; et al. DNA demethylation upregulated NRF2 expression in Alzheimer’s disease cellular model. Front. Aging Neurosci. 2016, 7, 244.
- Zhao, F.; Zhang, J.; Chang, N. Epigenetic modification of NRF2 by sulforaphane increases the antioxidative and anti-inflammatory capacity in a cellular model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 824, 1–10.
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of NRF2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111.
- Ba, Q.; Cui, C.; Wen, L.; Feng, S.; Zhou, J.; Yang, K. Schisandrin B shows neuroprotective effect in 6-OHDA-induced Parkinson’s disease via inhibiting the negative modulation of miR-34a on NRF2 pathway. Biomed Pharm. 2015, 75, 165–172.
- Wang, P.; Liang, X.; Lu, Y.; Zhao, X.; Liang, J. MicroRNA-93 downregulation ameliorates cerebral ischemic injury through the NRF2/HO-1 defense pathway. Neurochem. Res. 2016, 41, 2627–2635.
- Liu, P.; Zhao, H.; Wang, R.; Wang, P.; Tao, Z.; Gao, L.; Yan, F.; Liu, X.; Yu, S.; Ji, X.; et al. MicroRNA-424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke 2015, 46, 513–519.
- Dodson, M.; Zhang, D.D. Non-canonical activation of NRF2: New insights and its relevance to disease. Curr. Pathobiol. Rep. 2017, 5, 171–176.
- Fao, L.; Mota, S.I.; Rego, A.C. Shaping the NRF2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing. Res. Rev. 2019, 54, 100942.
- Yu, R.; Lei, W.; Mandlekar, S.; Weber, M.J.; Der, C.J.; Wu, J.; Kong, A.N. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J. Biol. Chem. 1999, 274, 27545–27552.
- Turjanski, A.G.; Vaque, J.P.; Gutkind, J.S. MAP kinases and the control of nuclear events. Oncogene 2007, 26, 3240–3253.
- Ainbinder, E.; Bergelson, S.; Pinkus, R.; Daniel, V. Regulatory mechanisms involved in activator-protein-1 (AP-1)-mediated activation of glutathione-S-transferase gene expression by chemical agents. Eur. J. Biochem. 1997, 243, 49–57.
- Fao, L.; Mota, S.I.; Rego, A.C. c-Src regulates NRF2 activity through PKCdelta after oxidant stimulus. Biochim. Biophys. Acta. Mol. Cell. Res. 2019, 1866, 686–698.
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631.
- Yue, J.; López, J.M. Understanding MAPK signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346.
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112.
- Yu, R.; Mandlekar, S.; Lei, W.; Fahl, W.E.; Tan, T.H.; Kong, A.N. P38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J. Biol. Chem. 2000, 275, 2322–2327.
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of NRF2 at multiple sites by MAP kinases has a limited contribution in modulating the NRF2-dependent antioxidant response. PLoS ONE 2009, 4, e6588.
- Prikas, E.; Poljak, A.; Ittner, A. Mapping p38α mitogen-activated protein kinase signaling by proximity-dependent labeling. Protein Sci. 2020, 29, 1196–1210.
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox. Signal 2006, 8, 1775–1789.
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877.
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405.
- Keum, Y.S.; Yu, S.; Chang, P.P.; Yuan, X.; Kim, J.H.; Xu, C.; Han, J.; Agarwal, A.; Kong, A.N. Mechanism of action of sulforaphane: Inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006, 66, 8804–8813.
- McNally, S.J.; Harrison, E.M.; Ross, J.A.; Garden, O.J.; Wigmore, S.J. Curcumin induces heme oxygenase 1 through generation of reactive oxygen species, p38 activation and phosphatase inhibition. Int. Int. J. Mol. Med. 2007, 19, 165–172.
- Rojo, A.I.; Medina-Campos, O.N.; Rada, P.; Zúñiga-Toala, A.; López-Gazcón, A.; Espada, S.; Pedraza-Chaverri, J.; Cuadrado, A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a KEAP1-independent regulation of NRF2 stability: Role of glycogen synthase kinase-3. Free Radic. Biol. Med. 2012, 52, 473–487.
- Sabapathy, K. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 145–169.
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell. Biol. 2007, 19, 142–149.
- Yamasaki, T.; Kawasaki, H.; Nishina, H. Diverse roles of JNK and MKK pathways in the brain. J. Signal Transduct. 2012, 459265.
- Thakur, A.; Wang, X.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Zhu, X. c-Jun phosphorylation in Alzheimer’s disease. J. Neurosci. Res. 2007, 85, 1668–1673.
- White, L.R.; Toft, M.; Kvam, S.N.; Farrer, M.J.; Aasly, J.O. MAPK-pathway activity, Lrrk2 G2019S, and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 1288–1294.
- Mitsios, N.; Gaffney, J.; Krupinski, J.; Mathias, R.; Wang, Q.; Hayward, S.; Rubio, F.; Kumar, P.; Kumar, S.; Slevin, M. Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem. Biophys. 2007, 47, 73–86.
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006, 131, 165–178.
- Chen, Y.; Liu, K.; Zhang, J.; Hai, Y.; Wang, P.; Wang, H.; Liu, Q.; Wong, C.C.; Yao, J.; Gao, Y.; et al. JNK phosphorylates the Neh6 domain of NRF2 and downregulates cytoprotective genes in acetaminophen-induced liver injury. Hepatology 2020, 71, 1787–1801.
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019, 35, 775–795.
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances NRF2-mediated transcriptional activation of the antioxidant response element. Degradation of NRF2 by the 26 S proteasome. Degradation of NRF2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541.
- Zipper, L.M.; Mulcahy, R.T. Inhibition of ERK and p38 MAP kinases inhibits binding of NRF2 and induction of GCS genes. Biochem. Biophys. Res. Commun. 2000, 278, 484–492.
- Yang, S.Y.; Pyo, M.C.; Nam, M.H.; Lee, K.W. ERK/NRF2 pathway activation by caffeic acid in HepG2 cells alleviates its hepatocellular damage caused by t-butylhydroperoxide-induced oxidative stress. BMC Complement. Altern. Med. 2019, 19, 139.
- Lee, D.S.; Kim, K.S.; Ko, W.; Li, B.; Jeong, G.S.; Jang, J.H.; Oh, H.; Kim, Y.C. The cytoprotective effect of sulfuretin against tert-butyl hydroperoxide-induced hepatotoxicity through NRF2/ARE and JNK/ERK MAPK-mediated heme oxygenase-1 expression. Int. J. Mol. Sci. 2014, 15, 8863–8877.
- Kitakaze, T.; Makiyama, A.; Samukawa, Y.; Jiang, S.; Yamashita, Y.; Ashida, H. A physiological concentration of luteolin induces phase II drug-metabolizing enzymes through the ERK1/2 signaling pathway in HepG2 cells. Arch. Biochem. Biophys. 2019, 663, 151–159.
- Yuan, X.; Xu, C.; Pan, Z.; Keum, Y.S.; Kim, J.H.; Shen, G.; Yu, S.; Oo, K.T.; Ma, J.; Kong, A.N. Butylated hydroxyanisole regulates ARE-mediated gene expression via NRF2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850.
- Mitani, T.; Yoshioka, Y.; Furuyashiki, T.; Yamashita, Y.; Shirai, Y.; Ashida, H. Enzymatically synthesized glycogen inhibits colitis through decreasing oxidative stress. Free Radic. Biol. Med. 2017, 106, 355–367.
- Steinberg, S.F. Mechanisms for redox-regulation of protein kinase C. Front. Pharm. 2015, 6, 128.
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of NRF2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774.
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of NRF2 at Ser40 by protein kinase C in response to antioxidants leads to the release of NRF2 from INRF2, but is not required for NRF2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J. Biol. Chem. 2003, 278, 44675–44682.
- Fang, X.; Yu, S.; Tanyi, J.L.; Lu, Y.; Woodgett, J.R.; Mills, G.B. Convergence of multiple signaling cascades at glycogen synthase kinase 3: EDG receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol. Cell. Biol. 2002, 22, 2099–2110.
- Wang, H.-Y.; Pisano, M.R.; Friedman, E. Attenuated protein kinase C activity and translocation in Alzheimer’s disease brain. Neurobiol. Aging 1994, 15, 293–298.
- Talman, V.; Pascale, A.; Jantti, M.; Amadio, M.; Tuominen, R.K. Protein Kinase C Activation as a potential therapeutic strategy in Alzheimer’s disease: Is there a role for embryonic lethal abnormal vision-like proteins? Basic Clin. Pharm. Toxicol. 2016, 119, 149–160.
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human NRF2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic. Biol. Med. 2007, 42, 1797–1806.
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of NRF2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of NRF2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76.
- Jain, A.K.; Jaiswal, A.K. Phosphorylation of tyrosine 568 controls nuclear export of NRF2. J. Biol. Chem. 2006, 281, 12132–12142.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Revisions:
3 times
(View History)
Update Date:
11 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No