 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael McKelvey | + 3095 word(s) | 3095 | 2021-05-12 08:43:26 | | | |
| 2 | Camila Xu | Meta information modification | 3095 | 2021-06-04 03:13:45 | | |
Video Upload Options
Proteases are enzymes that catalyse the hydrolysis of peptide bonds within proteins, facilitating their cleavage; this hydrolysis can either activate, inactivate, or modulate the activity of the target protein.
1. Introduction
Proteases are enzymes that catalyse the hydrolysis of peptide bonds within proteins, facilitating their cleavage; this hydrolysis can either activate, inactivate, or modulate the activity of the target protein. The identities of the amino acid residues that form the catalytic site have been used to group human proteases into serine, cysteine, matrix metallo-, aspartyl, and threonine protease classes. Within the lung, serine, cysteine and metalloproteases have received the most attention to date [1][2]. In healthy cells and tissues, both intracellular and extracellular protease activity is well managed by regulation at the transcriptional and translational levels, as well as by inhibitory pro-domains, modulatory factors (such as pH), and antiproteases at the protein level. However, higher-than-normal protease levels and excessive protease activity are recognised as hallmarks in chronic lung diseases (CLDs) and we continue to gain a greater appreciation of how the protease burden contributes to pathology [3][4][5].
Lung health is a product of many environmental and host factors, including exposure to toxins, particulates or pathogens, the mounting of appropriate immune responses to such stimuli, efficient ventilation mechanics and effective gas exchange. The mucosal surfaces of the airways are important interfaces for environmental and host factors, and alterations at this interface are a common feature in patients with CLD. The mucosal surface of the airway is composed of epithelial cells, many of which are ciliated, and is coated with a thin apical layer of mucus, resident and recruited immune cells, and the inhaled contents of the airway lumen. In many CLDs, the most obvious clinical symptoms are related to airway mucus, its excessive production, and an inability to clear it. MCC is a vital feature of the innate immune system in the airways [6][7]. A number of processes are essential to maintain effective MCC including regulation of ion channel activity, ciliary beat frequency (CBF), mucin expression and secretion and mucus viscosity [8]. Mucus is a hydrogel composed of water, salts, large mucin polymers, non-mucin proteins, lipids, and cellular debris [9][10]. Under normal conditions, water makes up 97–98% of mucus, producing a loose and mobile gel that ably protects the airway surface from inhaled pathogens and toxins, which are removed from the airways by ciliary beat and cough. However, in many CLDs, and especially the so-called ‘muco-obstructive’ lung diseases (chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), primary ciliary dyskinesia (PCD) and non-CF bronchiectasis), mucus composition is radically altered, producing a hyper-concentrated mucus layer [10][11][12][13]. The osmotic pressure of this hyper-concentrated mucus layer can exceed that of the subjacent periciliary layer, causing compression and flattening of the cilia, resulting in impaired ciliary beating and reduced mucus clearance. This leads to mucostasis and the build-up of mucus plaques and plugs in the airway lumen, producing muco-obstructive lung disease. The inciting causes of these original changes in the airways, mucus composition and MCC, vary between the different muco-obstructive lung diseases (environmental factors, recurrent infection, genetic mutations to ion channels etc.), but they share pathological mechanisms, many of which are mediated or modulated by proteases.
2. Proteases and Mucus
2.1. Proteases and Ion Transport
Ion channel activity is critical to maintain the airway surface liquid (ASL) at an appropriate height for effective MCC [14]. This is primarily achieved through the regulation of Cl− secretion and Na+ absorption via the chloride channel, cystic fibrosis transmembrane conductance regulator (CFTR) and the sodium channel, epithelial sodium channel (ENaC), respectively [15]. Defects in airway ion transport result in the development of muco-obstructive lung diseases, most notably with the loss of CFTR function in CF [16][17]. The role of proteases in regulating airway ion transport has largely focused on the activation of ENaC. ENaC undergoes maturation in the Golgi through the removal of an inhibitory peptide in its α-subunit by furin-type convertases [18]. These processed channels are classed as having intermediate open probability. However, release of a second inhibitory peptide from the γ-subunit at the plasma membrane can result in ENaC channels with a high open probability. This secondary cleavage is under the regulation of extracellular proteases. A number of proteases have been shown to cleave γ-ENaC, increasing the open probability of ENaC; these include serine proteases such as channel activating protease (CAP)-1, neutrophil elastase (NE), trypsin, chymotrypsin, prostasin and transmembrane protease serine 4 (TMPRSS4), as well as the cysteine proteases cathepsin B (CTSB) and cathepsin S (CTSS) [19][20][21][22][23][24]. Indeed, inhibition of trypsin-like serine proteases using the synthetic inhibitor ONO-3403 resulted in marked improvements in pulmonary dysfunction and emphysema in a murine model of CLD, indicating the importance of this ENaC-regulatory process [25]. Bacterial proteases including alkaline protease released from Pseudomonas aeruginosa also cleave and activate ENaC [26]. This activation of ENaC by both human and bacterial proteases is highly relevant in CLD, particularly where bacterial colonisation is prevalent. Increased ENaC activity is associated with severity in COPD and was shown to cause muco-obstructive lung disease in mice [27][28][29]. Conversely, decreased ENaC activity in patients with pseudohypoaldosteronism improved mucus clearance rates [30].
Protease-dependent regulation of CFTR has also been observed. Unlike ENaC, CFTR is not activated by proteolytic cleavage at the plasma membrane. However, the level of CFTR present at the cell surface is under the regulation of the cysteine protease calpain, which cleaves mature CFTR at the plasma membrane, allowing it to be internalised in vesicles for degradation [31]. Increased calpain activity is observed in CF, resulting in instability and reduced cell surface retention of CFTR that reaches the plasma membrane [32][33][34]. NE released from activated neutrophils, which are abundant in the chronically inflamed lung, also induces proteolysis and internalisation of CFTR on airway epithelial cells via the induction of this calpain-dependent degradation pathway [35]. Protease-dependent CFTR dysfunction may be important in chronic lung conditions beyond CF [36]. Indeed, CFTR function is associated with severity of emphysema in COPD [37]. There is also increasing evidence that loss of CFTR function resulting from exposure to cigarette smoke may promote smoking-associated lung disease [38][39]. These regulatory mechanisms are highly valuable in allowing dynamic changes in salt and water reabsorption and secretion in response to changing environments. However, in muco-obstructive lung disease, with a loss in protease/antiprotease balance, increased protease activity could lead to excessive Na+ absorption and/or loss of Cl− transport, with associated dehydration of the airways. Acidification of the ASL as a result of CFTR dysfunction may also play a part, stimulating the activity of cysteine cathepsins and further upregulating ENaC activity [40]. These studies highlight important roles for proteases in maintaining airway ion balance. Additionally, they suggest that targeting proteases may aid in regulating and maintaining effective ion transport and ASL height in muco-obstructive lung disease. The majority of research into protease regulation of airway ion channel activity has used cell culture models or Xenopus laevis oocytes. As such, there is currently little evidence for the direct therapeutic benefit of using protease inhibitors to alter ion channel activity in muco-obstructive lung disease, and this should be an area for future study.
2.2. Proteases and Ciliary Function
Cilia lining the epithelium of the airways play an important role in driving MCC; beating in a synchronised fashion, they facilitate removal of pathogens and debris trapped in the mucus layer. In the large airways, ciliated cells typically make up ~80% of the epithelium [41]. In muco-obstructive lung disease, ciliary beating is hindered by airway dehydration and increased mucus viscosity. Furthermore, as a result of goblet cell hyperplasia, the percentage of ciliated cells in the airway epithelium can drop as low as 20% [42]. The importance of proper ciliary function is evident in PCD, where abnormal ciliary beating leads to mucus plugging and chronic infection [43]. Protease activity contributes both directly and indirectly to the maintenance of ciliary stability and function. Optimal CBF is required for MCC and is regulated by a number of factors including cyclic adenosine monophosphate (cAMP)-dependent phosphorylation, intracellular Ca2+ levels and pH [44][45][46]. Ciliary beating is powered by molecular motors known as dyneins, which induce a series of contractions along the nine doublet microtubules making up the extracellular cilia axoneme and in doing so, produce the ciliary beat [47]. As such, dynein is an essential component of motile cilia. Cleavage of dynein by the serine proteases trypsin and subtilisin results in a loss of ciliary motility [48]. In addition to these human proteases, bacterial proteases are also capable of disrupting airway cilia by the same mechanism [48]. NE has also been shown to reduce CBF in vitro in human nasal bronchial epithelial cells. However, this effect was only observed in cells that were treated with high concentrations of NE [49]. Reductions in CBF in this case were likely a result of damage to the ciliated cells rather than a mechanistic alteration to ciliary beat, as histological examination revealed epithelial disruption while the number and ultrastructure of the cilia appeared normal [49]. This is still a significant finding because along with goblet cell metaplasia, this protease-dependent cell damage may contribute to the significant reduction in ciliated cells in the diseased airways. In contrast, while direct protease activity may lead to ciliated cell disruption, activation of protease-activated receptor (PAR)-2 by proteases secreted from airway neutrophils increases ciliary beating by 30–50% through the induction of Ca2+ signalling [50]. This could represent a clearance mechanism initiated during inflammation to clear inflammatory stimuli from the airways.
In addition to altering cilia motility, proteases can also affect cilia stability. Increased intracellular calpain activity is associated with diminished formation of cilia. Cilia require anchoring to the cell cytoskeleton by a basal body from which the axoneme is assembled. Calpain targets proteins in the basal body, resulting in a loss of anchoring and failed cilia formation [51]. The specific substrate(s) of calpain in the basal body structure have not been fully elucidated, though ezrin, a protein involved in plasma membrane/actin cytoskeleton interactions, is localised to the basal body and, as a known substrate of calpain, is a likely candidate [52]. These data present a varying effect of proteases on ciliary function. Increased protease activity in the chronically inflamed lung leads to reduced ciliary stability and motility and disruption of ciliated epithelial cells. Conversely, PAR-2 signalling may increase CBF in those ciliated cells that remain intact.
2.3. Proteases and Mucus Properties
Sitting atop the periciliary layer is a layer of mucus that traps debris and pathogens as it gradually moves from the distal to proximal airways along the mucociliary escalator. This mucus layer consists primarily of water, but also contains large polymeric mucin glycoproteins that determine the viscoelastic properties of the mucus layer [9]. These mucins are separated into secreted and tethered mucins depending on their properties. In the airways, the predominant secreted gel-forming mucins are mucin (MUC)-5AC and MUC5B [53]. Maintaining a mucus layer with the right properties is important for effective MCC and alterations in the composition of this mucus layer are associated with the development of chronic airway diseases [54][55][56]. Regulation is achieved through the maintenance of a number of factors including mucin expression and secretion, and mucus viscosity, which is largely determined by mucus hydration and crosslinking of mucins [10][57].
2.3.1. Mucin Expression
The role of proteases in the regulation of mucin gene expression has been examined in several studies, largely focusing on the regulation of MUC5AC expression, with little assessment of the regulation of MUC5B. This is likely a result of the current dogma that MUC5AC upregulation is the driving force behind mucus phenotypes in CLDs, while MUC5B is required for maintaining normal MCC [58]. The serine protease NE induces MUC5AC messenger ribonucleic acid (mRNA) and protein in airway epithelial cells (AECs) through increased mRNA stability or via a retinoic acid receptor-dependent mechanism [59][60]. Furthermore, induction of oxidative stress by NE has been shown to increase MUC5AC expression [61][62]. Changes in MUC5AC expression were not observed upon exposure of AECs to cysteine or metalloproteases in this study, suggesting these mechanisms may be specific to serine proteases [60]. However, in a separate study, a disintegrin and metalloprotease 17 (ADAM-17) and matrix metalloprotease 9 (MMP-9) induced MUC5AC expression through the activation of epidermal growth factor receptor (EGFR) [63]. Another serine protease, human airway trypsin-like protease (HAT) indirectly induced mucin gene expression in AECs through a similar mechanism [64]. Treatment of AECs with HAT induced expression and secretion of the EGFR ligand amphiregulin, leading to EGFR pathway activation and increased MUC5AC expression [64]. Interestingly, protease-mediated changed in CFTR and ENaC activity may also impact mucin production. For example, changes in these ion channels have been shown to lower intracellular Zn2+ concentrations by inducing alternative splicing of the zinc importer, ZIP2, which in turn drives MUC5AC hypersecretion [65].
In addition to human proteases, fungal proteases also regulate mucin expression. Notably, proteases released by Aspergillus fumigatus, a fungus that is highly prevalent in the early CF lung, induce MUC5AC expression [66][67]. A more recent study identified a Ras/Raf1/extracellular signal-regulated kinase (ERK) signalling pathway through which mucin expression was induced by fungal proteases [68]. Upregulation of MUC5AC by NE and other proteases in CLD will alter the MUC5AC/MUC5B ratio in favour of MUC5AC. This is important, as a higher MUC5AC/MUC5B ratio has been observed in pathogenic conditions including asthma [69]. The reason for the more pathogenic nature of MUC5AC is not fully understood. However, the tendency of MUC5AC to form sheets, and increased tethering to the airway epithelium, may play a part in impairing MCC to promote disease [70][71]. Impairing MCC would also be of benefit to fungal species trying to colonise the airway, giving an evolutionary advantage to those that induce MUC5AC expression. Future studies providing a clearer understanding of how proteases regulate the expression of MUC5B will be important not only in muco-obstructive lung disease, due to its role in MCC [58], but also the wider field of CLD including in idiopathic pulmonary fibrosis where a MUC5B promoter polymorphism and impaired MCC are associated with disease development [72][73].
2.3.2. Mucin Secretion
Following translation, mucins are packaged in a dehydrated form in secretory granules. Upon exocytosis the mucins are hydrated, absorbing more than 100 times their volume in water and, in the process, expand and acquire the correct viscoelastic properties to allow effective MCC [74]. Secretion of mucins is an incredibly rapid process occurring within a few hundred milliseconds [75]. Additionally, this secretory process is highly inducible, increasing over 1000-fold in response to certain stimuli [76][77]. Mucus hypersecretion is a major component of muco-obstructive lung diseases associated with declining lung function [78][79]. Metalloproteases including ADAM-10, meprin-α, and MMP-9, as well as the neutrophil serine proteases NE, cathepsin G and proteinase 3, are potent mucus secretagogues, inducing goblet cell degranulation and secretion of mucins from airway submucosal glands [80][81][82][83]. The specific mechanisms through which proteases induce mucin secretion are not fully understood. A number of key pathways have been highlighted in the literature. A study by Takeyama et al. demonstrated that cell-bound NE, but not free NE, could induce goblet cell degranulation, suggesting that a secondary signal may be required from the intercellular adhesion molecule (ICAM)-1 on the neutrophil cell surface to induce degranulation [84]. The intracellular signalling pathways that may be involved in this process were not elucidated in this study. More recently, NE was shown to induce mucin secretion via a protein kinase C (PKC)-dependent mechanism involving phosphorylation of myristoylated alanine-rich C kinase substrate (MARCKS), a PKC target and key regulator of mucin secretion [85]. Additionally, miR-146a negatively regulates NE-induced MUC5AC secretion from AECs through the inactivation of c-Jun N-terminal kinase (JNK) and nuclear factor kappa B (NF-κB) signaling [86]. Much like mucin expression, it is not only human proteases that regulate mucin secretion. Bacterial proteases including Pseudomonas elastase B, alkaline protease, and protease IV have all been shown to induce mucin secretion [87].
2.3.3. Mucus Viscoelastic Properties
Once secreted, gel-forming mucins MUC5AC and MUC5B form part of the mucus gel layer. The concentration of mucins in this layer contributes to its viscoelastic properties. Healthy mucus contains approximately 3% solids, having the consistency of egg whites [9]. However, in chronic lung disease this can increase to up to 15% solids as a result of airway dehydration coupled with increased mucin expression and hypersecretion [9]. However, it is not only the solid content of mucus that determines its viscoelastic properties; a number of other factors influence mucus viscosity including pH, extracellular deoxyribonucleic acid (DNA) content and the presence of mucin crosslinking, which occurs via the formation of disulphide bonds between mucins during oxidative stress [40][57][88]. Besides regulating mucin expression and secretion, proteases also regulate mucus viscoelastic properties by directly acting on secreted mucin proteins. In vitro studies have demonstrated that serine proteases are capable of degrading mucins [89]. While this would seem to suggest that protease activity may decrease mucus viscosity, this has not been directly measured. Importantly MUC5B is required for MCC and therefore its degradation could in fact hinder airway clearance [58]. Furthermore, proteases regulate the release of neutrophil extracellular traps (NETs) [90]. Induction of NET formation and subsequent increases in extracellular DNA may contribute to increased mucus viscosity. NETs also provide a protective lattice around proteases preventing access and inhibition by their natural inhibitors [91][92]. Bacterial species in the airway use mucolytic proteases to promote colonisation by inhibiting entrapment in the mucus layer and to gain access to the airway epithelium. P. aeruginosa-derived elastase B (pseudolysin) degrades both MUC5AC and MUC5B [89]. Mucins in the airways are highly sulphated, a mechanism to protect against degradation from bacterial proteases. However, P. aeruginosa has evolved the ability to secrete sulfatases, allowing it to bypass this protective barrier [93]. Fungal species including A. fumigatus break down mucins, not only to promote colonisation, but also to utilise it as a nutrient source [94]. A summary of the effects of proteases on mucus and MCC in muco-obstructive lung disease can be found in Figure 1.
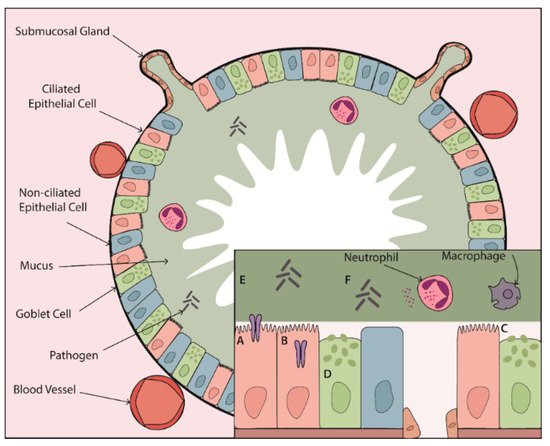
Figure 1. The effect of proteases on mucus and mucociliary clearance in the chronically inflamed airway. Proteases contribute to CLD pathogenesis through their impact on every step of the MCC mechanism. Elevated protease activity leads to (A) activation of ENaC and (B) loss of CFTR at the epithelial surface contributing to airway surface dehydration. (C) Protease-dependent damage to ciliated epithelial cells and cleavage of ciliary proteins leads to ineffective mucus clearance. This clearance defect is compounded by (D) protease-mediated increases in mucin expression and secretion from goblet cells and submucosal glands resulting in a highly viscous mucus layer that can no longer be cleared effectively. (E) Proteases can degrade mucins and (F) induce release of NETs, which may further alter mucus viscoelastic properties. Together, protease-dependent mucin/mucus hypersecretion and mucus dehydration produce highly viscous mucus, setting the stage for mucus plugging in the airways of patients with muco-obstructive lung disease.
References
- Kappelhoff, R.; Puente, X.S.; Wilson, C.H.; Seth, A.; López-Otín, C.; Overall, C.M. Overview of transcriptomic analysis of all human proteases, non-proteolytic homologs and inhibitors: Organ, tissue and ovarian cancer cell line expression profiling of the human protease degradome by the CLIP-CHIPTM DNA microarray. BBA-Mol. Cell Res. 2017, 1864, 2210–2219.
- Taggart, C.; Mall, M.A.; Lalmanach, G.; Cataldo, D.; Ludwig, A.; Janciauskiene, S.; Heath, N.; Meiners, S.; Overall, C.M.; Schultz, C.; et al. Protean proteases: At the cutting edge of lung diseases. Eur. Respir. J. 2017, 49, 1501200.
- Taggart, C.C.; Greene, C.M.; Carroll, T.P.; O’Neill, S.J.; McElvaney, N.G. Elastolytic proteases: Inflammation resolution and dysregu-lation in chronic infective lung disease. Am. J. Respir. Crit. Care Med. 2005, 171, 1070–1076.
- Greene, C.M.; McElvaney, N.G. Proteases and antiproteases in chronic neutrophilic lung disease-relevance to drug discovery. Br. J. Pharmacol. 2009, 158, 1048–1058.
- McKelvey, M.C.; Weldon, S.; McAuley, D.F.; Mall, M.A.; Taggart, C.C. Targeting Proteases in Cystic Fibrosis Lung Disease. Paradigms, Progress, and Potential. Am. J. Respir. Crit. Care Med. 2020, 201, 141–147.
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577.
- Mall, M.A. Role of Cilia, Mucus, and Airway Surface Liquid in Mucociliary Dysfunction: Lessons from Mouse Models. J. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 13–24.
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241.
- Fahy, J.V.; Dickey, B.F. Airway Mucus Function and Dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247.
- Boucher, R.C. Muco-Obstructive Lung Diseases. N. Engl. J. Med. 2019, 380, 1941–1953.
- Anderson, W.H.; Coakley, R.D.; Button, B.; Henderson, A.G.; Zeman, K.L.; Alexis, N.E.; Peden, D.B.; Lazarowski, E.R.; Davis, C.W.; Bailey, S.; et al. The Relationship of Mucus Concentration (Hydration) to Mucus Osmotic Pressure and Transport in Chronic Bronchitis. Am. J. Respir. Crit. Care Med. 2015, 192, 182–190.
- Button, B.; Anderson, W.H.; Boucher, R.C. Mucus Hyperconcentration as a Unifying Aspect of the Chronic Bronchitic Phenotype. Ann. Am. Thorac. Soc. 2016, 13, S156–S162.
- Henderson, A.G.; Ehre, C.; Button, B.; Abdullah, L.H.; Cai, L.-H.; Leigh, M.W.; DeMaria, G.C.; Matsui, H.; Donaldson, S.H.; Davis, C.W.; et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Investig. 2014, 124, 3047–3060.
- Tarran, R. Regulation of Airway Surface Liquid Volume and Mucus Transport by Active Ion Transport. Proc. Am. Thorac. Soc. 2004, 1, 42–46.
- Tarran, R.; Button, B.; Picher, M.; Paradiso, A.M.; Ribeiro, C.M.; Lazarowski, E.R.; Zhang, L.; Collins, P.L.; Pickles, R.J.; Fredberg, J.J.; et al. Normal and cystic fibrosis airway surface liquid homeostasis: The effects of phasic shear stress and viral infections. J. Biol. Chem. 2005, 280, 35751–35759.
- Kunzelmann, K.; Greger, R. Na+ and Cl− conductances in airway epithelial cells: Increased Na+ conductance in cystic fibrosis. Pflügers Archiv-Eur. J. Physiol. 1995, 431, 4371.
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073.
- Hughey, R.P.; Bruns, J.B.; Kinlough, C.L.; Harkleroad, K.L.; Tong, Q.; Carattino, M.D.; Johnson, J.P.; Stockand, J.D.; Kleyman, T.R. Epithelial Sodium Channels Are Activated by Furin-dependent Proteolysis. J. Biol. Chem. 2004, 279, 18111–18114.
- Bruns, J.B.; Carattino, M.D.; Sheng, S.; Maarouf, A.B.; Weisz, O.A.; Pilewski, J.M.; Hughey, R.P.; Kleyman, T.R. Epithelial Na+ Channels Are Fully Activated by Furin- and Prostasin-dependent Release of an Inhibitory Peptide from the γ-Subunit. J. Biol. Chem. 2007, 282, 6153–6160.
- Haerteis, S.; Krappitz, M.; Bertog, M.; Krappitz, A.; Baraznenok, V.; Henderson, I.; Lindström, E.; Murphy, J.E.; Bunnett, N.W.; Korbmacher, C. Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflügers Archiv-Eur. J. Physiol. 2012, 464, 353–365.
- García-Caballero, A.; Dang, Y.; He, H.; Stutts, M.J. ENaC Proteolytic Regulation by Channel-activating Protease 2. J. Gen. Physiol. 2008, 132, 521–535.
- Caldwell, R.A.; Boucher, R.C.; Stutts, M.J. Serine protease activation of near-silent epithelial Na+ channels. Am. J. Physiol. Physiol. 2004, 286, C190–C194.
- Larionov, A.; Dahlke, E.; Kunke, M.; Rodriguez, L.Z.; Schiessl, I.M.; Magnin, J.; Kern, U.; Alli, A.A.; Mollet, G.; Schilling, O.; et al. Cathepsin B increases ENaC activity leading to hypertension early in nephrotic syndrome. J. Cell. Mol. Med. 2019, 23, 6543–6553.
- Caldwell, R.A.; Boucher, R.C.; Stutts, M.J. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am. J. Physiol. Cell. Mol. Physiol. 2005, 288, L813–L819.
- Shuto, T.; Kamei, S.; Nohara, H.; Fujikawa, H.; Tasaki, Y.; Sugahara, T.; Ono, T.; Matsumoto, C.; Sakaguchi, Y.; Maruta, K.; et al. Pharmacological and genetic reappraisals of protease and oxidative stress pathways in a mouse model of obstructive lung diseases. Sci. Rep. 2016, 6, 39305.
- Butterworth, M.B.; Zhang, L.; Liu, X.; Shanks, R.M.; Thibodeau, P.H. Modulation of the Epithelial Sodium Channel (ENaC) by Bacterial Metalloproteases and Protease Inhibitors. PLoS ONE 2014, 9, e100313.
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O’Neal, W.K.; Boucher, R.C.; Neal, W.K.O. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493.
- Zhao, R.; Liang, X.; Zhao, M.; Liu, S.-L.; Huang, Y.; Idell, S.; Li, X.; Ji, H.-L. Correlation of Apical Fluid-Regulating Channel Proteins with Lung Function in Human COPD Lungs. PLoS ONE 2014, 9, e109725.
- Mall, M.A.; Harkema, J.R.; Trojanek, J.B.; Treis, D.; Livraghi, A.; Schubert, S.; Zhou, Z.; Kreda, S.M.; Tilley, S.L.; Hudson, E.J.; et al. Development of chronic bronchitis and emphysema in β-epithelial Na+ channel-overexpressing mice. Am. J. Respir. Crit. Care Med. 2008, 177, 730–742.
- Kerem, E.; Bistritzer, T.; Hanukoglu, A.; Hofmann, T.; Zhou, Z.; Bennett, W.; MacLaughlin, E.; Barker, P.; Nash, M.; Quittell, L.; et al. Pulmonary Epithelial Sodium-Channel Dysfunction and Excess Airway Liquid in Pseudohypoaldosteronism. N. Engl. J. Med. 1999, 341, 156–162.
- Averna, M.; Stifanese, R.; Grosso, R.; Pedrazzi, M.; De Tullio, R.; Salamino, F.; Pontremoli, S.; Melloni, E. Role of calpain in the regulation of CFTR (cystic fibrosis transmembrane conductance regulator) turnover. Biochem. J. 2010, 430, 255–263.
- Matos, A.M.; Pinto, F.R.; Barros, P.; Amaral, M.D.; Pepperkok, R.; Matos, P. Inhibition of calpain 1 restores plasma membrane stability to pharmacologically rescued Phe508del-CFTR variant. J. Biol. Chem. 2019, 294, 13396–13410.
- Averna, M.; Stifanese, R.; De Tullio, R.; Minicucci, L.; Cresta, F.; Palena, S.; Salamino, F.; Pontremoli, S.; Melloni, E. Evidence for alteration of calpain/calpastatin system in PBMC of cystic fibrosis patients. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1649–1657.
- Averna, M.; Pedrazzi, M.; Minicucci, L.; De Tullio, R.; Cresta, F.; Salamino, F.; Pontremoli, S.; Melloni, E. Calpain Inhibition Promotes the Rescue of F508del-CFTR in PBMC from Cystic Fibrosis Patients. PLoS ONE 2013, 8, e66089.
- Le Gars, M.; Descamps, D.; Roussel, D.; Saussereau, E.; Guillot, L.; Ruffin, M.; Tabary, O.; Hong, S.S.; Boulanger, P.; Paulais, M.; et al. Neutrophil elastase degrades cystic fibrosis transmembrane conductance regulator via calpains and disables channel function in vitro and in vivo. Am. J. Respir. Crit. Care Med. 2013, 187, 170–179.
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054.
- Bodas, M.; Min, T.; Mazur, S.; Vij, N. Critical Modifier Role of Membrane-Cystic Fibrosis Transmembrane Conductance Regula-tor-Dependent Ceramide Signaling in Lung Injury and Emphysema. J. Immunol. 2011, 186, 602–613.
- Cantin, A.M.; Hanrahan, J.W.; Bilodeau, G.; Ellis, L.; Dupuis, A.; Liao, J.; Zielenski, J.; Durie, P. Cystic Fibrosis Transmembrane Conductance Regulator Function Is Suppressed in Cigarette Smokers. Am. J. Respir. Crit. Care Med. 2006, 173, 1139–1144.
- Clunes, L.A.; Davies, C.M.; Coakley, R.D.; Aleksandrov, A.A.; Henderson, A.G.; Zeman, K.L.; Worthington, E.N.; Gentzsch, M.; Kreda, S.M.; Cholon, D.; et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2011, 26, 533–545.
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.v.E.M.; Bánfi, B.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113.
- Haitchi, H.M.; Yoshisue, H.; Ribbene, A.; Wilson, S.J.; Holloway, J.W.; Bucchieri, F.; Hanley, N.A.; Wilson, D.I.; Zummo, G.; Holgate, S.T.; et al. Chronological expression of Ciliated Bronchial Epithelium 1 during pulmonary development. Eur. Respir. J. 2009, 33, 1095–1104.
- Davis, C.W.; Dickey, B.F. Regulated Airway Goblet Cell Mucin Secretion. Annu. Rev. Physiol. 2008, 70, 487–512.
- Lucas, J.S.; Burgess, A.; Mitchison, H.M.; Moya, E.; Williamson, M.; Hogg, C.; on behalf of the National PCD Service, UK. Diagnosis and management of primary ciliary dyskinesia. Arch. Dis. Child. 2014, 99, 850–856.
- Wyatt, T.A.; Spurzem, J.R.; May, K.; Sisson, J.H. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 1998, 275, L827–L835.
- Di Benedetto, G.; Magnus, C.J.; Gray, P.T.; Mehta, A. Calcium regulation of ciliary beat frequency in human respiratory epithelium in vitro. J. Physiol. 1991, 439, 103–113.
- Sutto, Z.; Conner, G.E.; Salathe, M. Regulation of human airway ciliary beat frequency by intracellular pH. J. Physiol. 2004, 560, 519–532.
- Satir, P.; Heuser, T.; Sale, W.S. A Structural Basis for How Motile Cilia Beat. Bioscience 2014, 64, 1073–1083.
- Hingley, S.T.; Hastie, A.T.; Kueppers, F.; Higgins, M.L. Disruption of respiratory cilia by proteases including those of Pseudomonas aeruginosa. Infect. Immun. 1986, 54, 379–383.
- Amitani, R.; Wilson, R.; Rutman, A.; Read, R.; Ward, C.; Burnett, D.; Stockley, R.A.; Cole, P.J. Effects of Human Neutrophil Elastase andPseudomonas aeruginosaProteinases on Human Respiratory Epithelium. Am. J. Respir. Cell Mol. Biol. 1991, 4, 26–32.
- McMahon, D.B.; Workman, A.D.; Kohanski, M.A.; Carey, R.M.; Freund, J.R.; Hariri, B.M.; Chen, B.; Doghramji, L.J.; Adappa, N.D.; Palmer, J.N.; et al. Protease-activated receptor 2 activates airway apical membrane chloride permeability and increases ciliary beating. FASEB J. 2018, 32, 155–167.
- Gomperts, B.N.; Gong-Cooper, X.; Hackett, B.P. Foxj1 regulates basal body anchoring to the cytoskeleton of ciliated pulmonary epithelial cells. J. Cell Sci. 2004, 117, 1329–1337.
- Roberts, R.E.; Martin, M.; Marion, S.; Elumalai, G.L.; Lewis, K.; Hallett, M.B. Ca2+-activated cleavage of ezrin visualised dynamically in living myeloid cells during cell surface area expansion. J. Cell Sci. 2020, 133.
- Kirkham, S.; Sheehan, J.K.; Knight, D.; Richardson, P.S.; Thornton, D.J. Heterogeneity of airways mucus: Variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem. J. 2002, 361, 537–546.
- Hogg, J.C.; Chu, F.S.; Tan, W.C.; Sin, D.D.; Patel, S.A.; Pare, P.D.; Martinez, F.J.; Rogers, R.M.; Make, B.J.; Criner, G.J.; et al. Survival after lung volume reduction in chronic obstructive pulmonary disease: Insights from small airway pathology. Am. J. Respir. Crit. Care Med. 2007, 176, 454–459.
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur. Respir. J. 2018, 52, 1801297.
- Ramsey, K.A.; Chen, A.C.H.; Radicioni, G.; Lourie, R.; Martin, M.; Broomfield, A.; Sheng, Y.H.; Hasnain, S.Z.; Radford-Smith, G.; Simms, L.A.; et al. Airway Mucus Hyperconcentration in Non–Cystic Fibrosis Bronchiectasis. Am. J. Respir. Crit. Care Med. 2020, 201, 661–670.
- Yuan, S.; Hollinger, M.; Lachowicz-Scroggins, M.E.; Kerr, S.C.; Dunican, E.M.; Daniel, B.M.; Ghosh, S.; Erzurum, S.C.; Willard, B.; Hazen, S.L.; et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci. Transl. Med. 2015, 7, 276ra27.
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nat. Cell Biol. 2014, 505, 412–416.
- Ja, S.K.; Kim, Y.D.; Jetten, A.M.; Belloni, P.; Nettesheim, P. Overexpression of mucin genes induced by interleukin-1β, tumor necrosis factor-α, lipopolysaccharide, and neutrophil elastase is inhibited by a retinoic acid receptor α antagonist. Exp. Lung Res. 2002, 28, 315–332.
- Voynow, J.A.; Rosenthal Young, L.; Wang, Y.; Horger, T.; Rose, M.C.; Fischer, B.M. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L835–L843.
- Fischer, B.; Voynow, J. Neutrophil elastase induces MUC5AC messenger RNA expression by an oxidant-dependent mecha-nism. Chest 2000, 117, 317S–320S.
- Shao, M.X.G.; Nadel, J.A. Neutrophil Elastase Induces MUC5AC Mucin Production in Human Airway Epithelial Cells via a Cascade Involving Protein Kinase C, Reactive Oxygen Species, and TNF-α-Converting Enzyme. J. Immunol. 2005, 175, 4009–4016.
- Deshmukh, H.S.; Case, L.M.; Wesselkamper, S.C.; Borchers, M.T.; Martin, L.D.; Shertzer, H.G.; Nadel, J.A.; Leikauf, G.D. Metalloproteinases Mediate Mucin 5AC Expression by Epidermal Growth Factor Receptor Activation. Am. J. Respir. Crit. Care Med. 2005, 171, 305–314.
- Chokki, M.; Yamamura, S.; Eguchi, H.; Masegi, T.; Horiuchi, H.; Tanabe, H.; Kamimura, T.; Yasuoka, S. Human Airway Trypsin-Like Protease Increases Mucin Gene Expression in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 470–478.
- Kamei, S.; Fujikawa, H.; Nohara, H.; Ueno-Shuto, K.; Maruta, K.; Nakashima, R.; Kawakami, T.; Matsumoto, C.; Sakaguchi, Y.; Ono, T.; et al. Zinc Deficiency via a Splice Switch in Zinc Importer ZIP2/SLC39A2 Causes Cystic Fibrosis-Associated MUC5AC Hypersecretion in Airway Epithelial Cells. EBioMedicine 2018, 27, 304–316.
- Oguma, T.; Asano, K.; Tomomatsu, K.; Kodama, M.; Fukunaga, K.; Shiomi, T.; Ohmori, N.; Ueda, S.; Takihara, T.; Shiraishi, Y.; et al. Induction of Mucin and MUC5AC Expression by the Protease Activity of Aspergillus fumigatus in Airway Epithelial Cells. J. Immunol. 2011, 187, 999–1005.
- Saunders, R.V.; Modha, D.E.; Claydon, A.; Gaillard, E.A. Chronic Aspergillus fumigatus colonization of the pediatric cystic fibrosis airway is common and may be associated with a more rapid decline in lung function. Med. Mycol. 2016, 54, 537–543.
- Wu, X.; Lee, B.; Zhu, L.; Ding, Z.; Chen, Y. Exposure to mold proteases stimulates mucin production in airway epithelial cells through Ras/Raf1/ERK signal pathway. PLoS ONE 2020, 15, e0231990.
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Arron, J.R.; Koth, L.L.; Fahy, J.V. T-helper Type 2–driven Inflammation Defines Major Subphenotypes of Asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395.
- Bonser, L.R.; Zlock, L.; Finkbeiner, W.; Erle, D.J. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J. Clin. Investig. 2016, 126, 2367–2371.
- Ostedgaard, L.S.; Moninger, T.O.; McMenimen, J.D.; Sawin, N.M.; Parker, C.P.; Thornell, I.M.; Powers, L.S.; Gansemer, N.D.; Bouzek, D.C.; Cook, D.P.; et al. Gel-forming mucins form distinct morphologic structures in airways. Proc. Natl. Acad. Sci. USA 2017, 114, 6842–6847.
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512.
- Duerr, J.; Leitz, D.H.W.; Szczygiel, M.; Dvornikov, D.; Fraumann, S.G.; Kreutz, C.; Zadora, P.K.; Agircan, A.S.; Konietzke, P.; Engelmann, T.A.; et al. Conditional deletion of Nedd4–2 in lung epi-thelial cells causes progressive pulmonary fibrosis in adult mice. Nat. Commun. 2020, 11, 2012.
- Jaramillo, A.M.; Azzegagh, Z.; Tuvim, M.J.; Dickey, B.F. Airway Mucin Secretion. Ann. Am. Thorac. Soc. 2018, 15, S164–S170.
- Shumilov, D.; Popov, A.; Fudala, R.; Akopova, I.; Gryczynski, I.; Borejdo, J.; Gryczynski, Z.; Grygorczyk, R. Real-time imaging of exocytotic mucin release and swelling in Calu-3 cells using acridine orange. Methods 2014, 66, 312–324.
- Davis, C.W.; Dowell, M.L.; Lethem, M.; Van Scott, M. Goblet cell degranulation in isolated canine tracheal epithelium: Response to exogenous ATP, ADP, and adenosine. Am. J. Physiol. Cell Physiol. 1992, 262, C1313–C1323.
- Lethem, M.I.; Dowell, M.L.; Van Scott, M.; Yankaskas, J.R.; Egan, T.; Boucher, R.C.; Davis, C.W. Nucleotide Regulation of Goblet Cells in Human Airway Epithelial Explants: Normal Exocytosis in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 1993, 9, 315–322.
- Vestbo, J.; Prescott, E.; Lange, P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Am. J. Respir. Crit. Care Med. 1996, 153, 1530–1535.
- Martínez-Rivera, C.; Crespo, A.; Pinedo-Sierra, C.; García-Rivero, J.L.; Pallarés-Sanmartín, A.; Marina-Malanda, N.; Pascual-Erquicia, S.; Padilla, A.; Mayoralas-Alises, S.; Plaza, V.; et al. Mucus hypersecretion in asthma is associated with rhinosinusitis, polyps and exacerbations. Respir. Med. 2018, 135, 22–28.
- Lemjabbar, H.; Basbaum, C. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat. Med. 2002, 8, 41–46.
- Nadel, J.A. Role of Mast Cell and Neutrophil Proteases in Airway Secretion. Am. Rev. Respir. Dis. 1991, 144, S48–S51.
- Lundgren, J.D.; Rieves, R.D.; Mullol, J.; Logun, C.; Shelhamer, J.H. The effect of neutrophil proteinase enzymes on the release of mucus from feline and human airway cultures. Respir. Med. 1994, 88, 511–518.
- Witko-Sarsat, V.; Halbwachs-Mecarelli, L.; Schuster, A.; Nusbaum, P.; Ueki, I.; Canteloup, S.; Lenoir, G.; Descamps-Latscha, B.; Nadel, J.A. Proteinase 3, a Potent Secretagogue in Airways, Is Present in Cystic Fibrosis Sputum. Am. J. Respir. Cell Mol. Biol. 1999, 20, 729–736.
- Takeyama, K.; Agustí, C.; Ueki, I.; Lausier, J.; Cardell, L.O.; Nadel, J.A. Neutrophil-dependent goblet cell degranulation: Role of membrane-bound elastase and adhesion molecules. Am. J. Physiol. Content 1998, 275, L294–L302.
- Park, J.-A.; He, F.; Martin, L.D.; Li, Y.; Chorley, B.N.; Adler, K.B. Human Neutrophil Elastase Induces Hypersecretion of Mucin from Well-Differentiated Human Bronchial Epithelial Cells in Vitro via a Protein Kinase Cδ-Mediated Mechanism. Am. J. Pathol. 2005, 167, 651–661.
- Zhong, T.; Perelman, J.M.; Kolosov, V.P.; Zhou, X.-D. MiR-146a negatively regulates neutrophil elastase-induced MUC5AC secretion from 16HBE human bronchial epithelial cells. Mol. Cell. Biochem. 2011, 358, 249–255.
- Klinger, J.D.; Tandler, B.; Liedtke, C.M.; Boat, T.F. Proteinases of Pseudomonas aeruginosa evoke mucin release by tracheal epithe-lium. J. Clin. Investig. 1984, 74, 1669–1678.
- Puchelle, E.; Zahm, J.-M.; De Bentzmann, S.; Grosskopf, C.; Shak, S.; Mougel, D.; Polu, J.-M. Effects of rhDNase on purulent airway secretions in chronic bronchitis. Eur. Respir. J. 1996, 9, 765–769.
- Henke, M.O.; John, G.; Rheineck, C.; Chillappagari, S.; Naehrlich, L.; Rubin, B.K. Serine Proteases Degrade Airway Mucins in Cystic Fibrosis. In Proceedings of the American Thoracic Society 2011 International Conference, Denver, CO, USA, 13–18 May 2011; Volume 79, pp. 3438–3444.
- Majewski, P.; Majchrzak-Gorecka, M.; Grygier, B.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Cichy, J. Inhibitors of Serine Proteases in Regulating the Production and Function of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 261.
- Dubois, A.V.; Gauthier, A.; Bréa, D.; Varaigne, F.; Diot, P.; Gauthier, F.; Attucci, S. Influence of DNA on the Activities and Inhibition of Neutrophil Serine Proteases in Cystic Fibrosis Sputum. Am. J. Respir. Cell Mol. Biol. 2012, 47, 80–86.
- Guerra, M.; Halls, V.S.; Schatterny, J.; Hagner, M.; Mall, M.A.; Schultz, C. Protease FRET Reporters Targeting Neutrophil Extracellular Traps. J. Am. Chem. Soc. 2020, 142, 20299–20305.
- Robinson, C.V.; Elkins, M.R.; Bialkowski, K.M.; Thornton, D.J.; Kertesz, M.A. Desulfurization of mucin by Pseudomonas aeruginosa: Influence of sulfate in the lungs of cystic fibrosis patients. J. Med. Microbiol. 2012, 61, 1644–1653.
- Cowley, A.C.; Thornton, D.J.; Denning, D.W.; Horsley, A. Aspergillosis and the role of mucins in cystic fibrosis. Pediatr. Pulmonol. 2016, 52, 548–555.

