 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Willianne Hoepel | + 6200 word(s) | 6200 | 2021-05-17 08:55:20 | | | |
| 2 | Vivi Li | Meta information modification | 6200 | 2021-06-04 03:15:33 | | |
Video Upload Options
Macrophages play a key role in induction of inflammatory responses. These inflammatory responses are mostly considered to be instigated by activation of pattern recognition receptors (PRRs) or cytokine receptors. However, recently it has become clear that also antibodies and pentraxins, which can both activate Fc receptors (FcRs), induce very powerful inflammatory responses by macrophages that can even be an order of magnitude greater than PRRs. While the physiological function of this antibody-dependent inflammation (ADI) is to counteract infections, undesired activation or over-activation of this mechanism will lead to pathology, as observed in a variety of disorders, including viral infections such as COVID-19, chronic inflammatory disorders such as Crohn’s disease, and autoimmune diseases such as rheumatoid arthritis.
1. Introduction
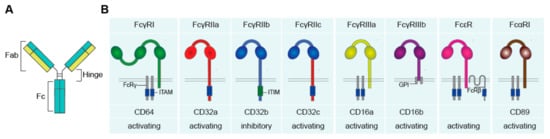
Antibodies and Fc Receptors
2. Physiological Immune Activation: Host Defense against Pathogens
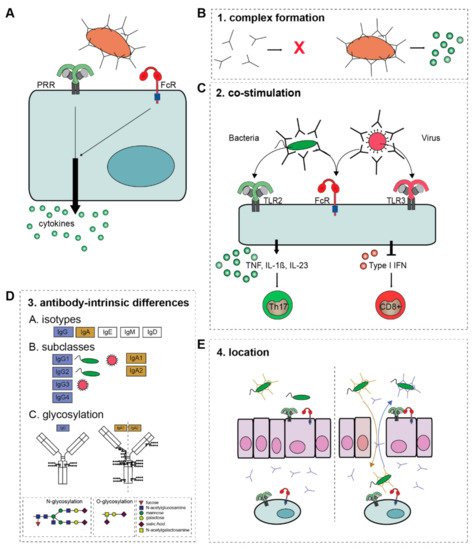
2.1. Molecular Mechanism of Antibody-Induced Inflammation
2.1.1. Complex Formation
2.1.2. Co-Stimulation
2.1.3. Antibody-Intrinsic Differences
Isotype
2.1.4. Location
2.2. Inflammation by Pentraxins
2.2.1. Pentraxin Family
2.2.2. Pentraxin-Induced Inflammation
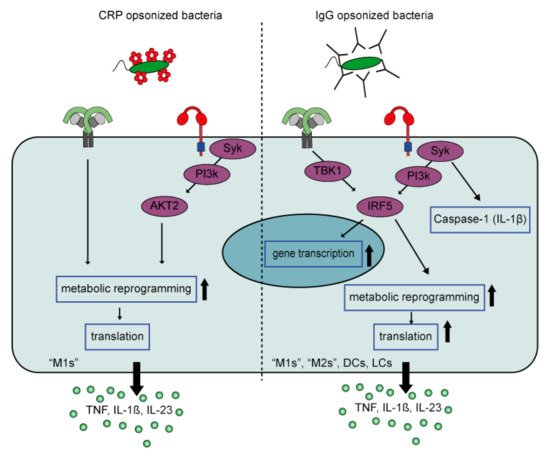
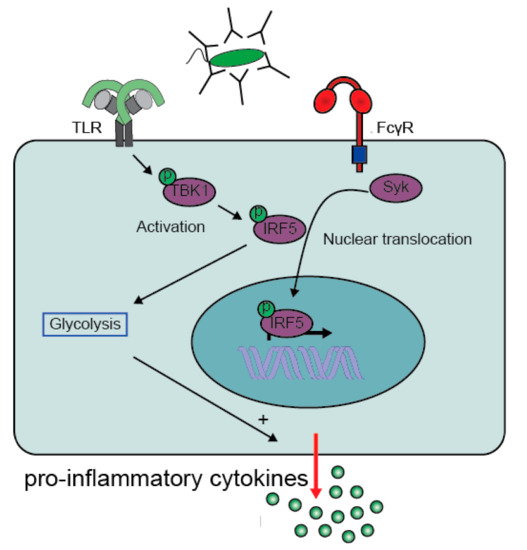
3. Pathological Immune Activation
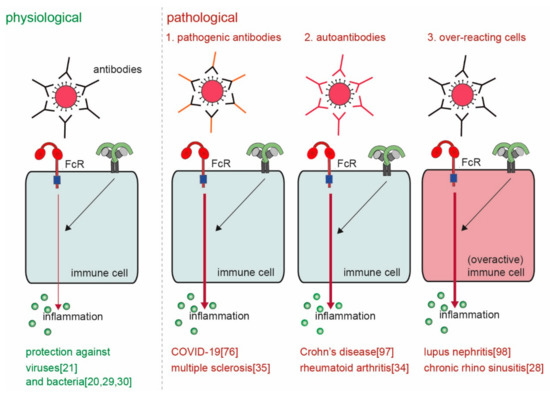
3.1. Pathogenic Antibodies
3.2. Autoantibodies
3.3. Cell-Intrinsic Overactivation
3.4. Aberrant Location/Concentration
References
- Van Egmond, M.; Vidarsson, G.; Bakema, J.E. Cross-talk between pathogen recognizing Toll-like receptors and immunoglobulin Fc receptors in immunity. Immunol. Rev. 2015, 268, 311–327.
- Kieser, K.J.; Kagan, J.C. Multi-receptor detection of individual bacterial products by the innate immune system. Nat. Rev. Immunol. 2017, 17, 376–390.
- Geijtenbeek, T.B.H.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479.
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066.
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond binding: Antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018, 18, 46–61.
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520.
- Van Erp, E.A.; Luytjes, W.; Ferwerda, G.; van Kasteren, P.B. Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease. Front. Immunol. 2019, 10, 548.
- Kapur, R.; Einarsdottir, H.K.; Vidarsson, G. IgG-effector functions: "the good, the bad and the ugly". Immunol. Lett. 2014, 160, 139–144.
- De Taeye, S.W.; Rispens, T.; Vidarsson, G. The Ligands for Human IgG and Their Effector Functions. Antibodies 2019, 8, 30.
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47.
- Nimmerjahn, F.; Gordan, S.; Lux, A. FcgammaR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol. 2015, 36, 325–336.
- Getahun, A.; Cambier, J.C. Of ITIMs, ITAMs, and ITAMis: Revisiting immunoglobulin Fc receptor signaling. Immunol. Rev. 2015, 268, 66–73.
- van der Heijden, J.; Breunis, W.B.; Geissler, J.; de Boer, M.; van den Berg, T.K.; Kuijpers, T.W. Phenotypic Variation in IgG Receptors by Nonclassical FCGR2C Alleles. J. Immunol. 2012, 188, 1318.
- Bruhns, P.; Jönsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51.
- Pasquier, B.; Launay, P.; Kanamaru, Y.; Moura, I.C.; Pfirsch, S.; Ruffié, C.; Hénin, D.; Benhamou, M.; Pretolani, M.; Blank, U.; et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: Dual role of FcRgamma ITAM. Immunity 2005, 22, 31–42.
- Stapleton, N.M.; Einarsdóttir, H.K.; Stemerding, A.M.; Vidarsson, G. The multiple facets of FcRn in immunity. Immunol. Rev. 2015, 268, 253–268.
- Stapleton, N.M.; Andersen, J.T.; Stemerding, A.M.; Bjarnarson, S.P.; Verheul, R.C.; Gerritsen, J.; Zhao, Y.; Kleijer, M.; Sandlie, I.; de Haas, M.; et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat. Commun. 2011, 2, 599.
- Vidarsson, G.; Stemerding, A.M.; Stapleton, N.M.; Spliethoff, S.E.; Janssen, H.; Rebers, F.E.; de Haas, M.; van de Winkel, J.G. FcRn: An IgG receptor on phagocytes with a novel role in phagocytosis. Blood 2006, 108, 3573–3579.
- Rhodes, D.A.; Isenberg, D.A. TRIM21 and the Function of Antibodies inside Cells. Trends Immunol. 2017, 38, 916–926.
- den Dunnen, J.; Vogelpoel, L.T.; Wypych, T.; Muller, F.J.; de Boer, L.; Kuijpers, T.W.; Zaat, S.A.; Kapsenberg, M.L.; de Jong, E.C. IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcgammaRIIa in human dendritic cells. Blood 2012, 120, 112–121.
- Newling, M.; Hoepel, W.; Vogelpoel, L.T.C.; Heineke, M.H.; van Burgsteden, J.A.; Taanman-Kueter, E.W.M.; Eggink, D.; Kuijpers, T.W.; Beaumont, T.; van Egmond, M.; et al. Fc gamma receptor IIa suppresses type I and III interferon production by human myeloid immune cells. Eur. J. Immunol. 2018, 48, 1796–1809.
- Vogelpoel, L.T.; Baeten, D.L.; de Jong, E.C.; den Dunnen, J. Control of cytokine production by human fc gamma receptors: Implications for pathogen defense and autoimmunity. Front. Immunol. 2015, 6, 79.
- Ben Mkaddem, S.; Benhamou, M.; Monteiro, R.C. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front. Immunol. 2019, 10, 811.
- Chauhan, A.K. Human CD4+ T-Cells: A Role for Low-Affinity Fc Receptors. Front. Immunol. 2016, 7, 215.
- Wang, Y.; Jönsson, F. Expression, Role, and Regulation of Neutrophil Fcγ Receptors. Front. Immunol. 2019, 10, 1958.
- Devaraj, S.; Du Clos Terry, W.; Jialal, I. Binding and Internalization of C-Reactive Protein by Fcgamma Receptors on Human Aortic Endothelial Cells Mediates Biological Effects. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1359–1363.
- Cheeseman, H.M.; Carias, A.M.; Evans, A.B.; Olejniczak, N.J.; Ziprin, P.; King, D.F.L.; Hope, T.J.; Shattock, R.J. Expression Profile of Human Fc Receptors in Mucosal Tissue: Implications for Antibody-Dependent Cellular Effector Functions Targeting HIV-1 Transmission. PLoS ONE 2016, 11, e0154656.
- Golebski, K.; Hoepel, W.; van Egmond, D.; de Groot, E.J.; Amatngalim, G.D.; Beekman, J.M.; Fokkens, W.J.; van Drunen, C.M.; den Dunnen, J. FcgammaRIII stimulation breaks the tolerance of human nasal epithelial cells to bacteria through cross-talk with TLR4. Mucosal Immunol. 2019, 12, 425–433.
- Hoepel, W.; Newling, M.; Vogelpoel, L.T.C.; Sritharan, L.; Hansen, I.S.; Kapsenberg, M.L.; Baeten, D.L.P.; Everts, B.; den Dunnen, J. FcgammaR-TLR Cross-Talk Enhances TNF Production by Human Monocyte-Derived DCs via IRF5-Dependent Gene Transcription and Glycolytic Reprogramming. Front. Immunol. 2019, 10, 739.
- Vogelpoel, L.T.; Hansen, I.S.; Visser, M.W.; Nagelkerke, S.Q.; Kuijpers, T.W.; Kapsenberg, M.L.; de Jong, E.C.; den Dunnen, J. FcgammaRIIa cross-talk with TLRs, IL-1R, and IFNgammaR selectively modulates cytokine production in human myeloid cells. Immunobiology 2015, 220, 193–199.
- Hansen, I.S.; Hoepel, W.; Zaat, S.A.J.; Baeten, D.L.P.; den Dunnen, J. Serum IgA Immune Complexes Promote Proinflammatory Cytokine Production by Human Macrophages, Monocytes, and Kupffer Cells through FcalphaRI-TLR Cross-Talk. J. Immunol. 2017, 199, 4124–4131.
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheumatol. 2011, 63, 53–62.
- Bakema, J.E.; Tuk, C.W.; van Vliet, S.J.; Bruijns, S.C.; Vos, J.B.; Letsiou, S.; Dijkstra, C.D.; van, K.Y.; Brenkman, A.B.; Van Egmond, M. Antibody-opsonized bacteria evoke an inflammatory dendritic cell phenotype and polyfunctional Th cells by cross-talk between TLRs and FcRs. J. Immunol. 2015, 194, 1856–1866.
- Vogelpoel, L.T.; Hansen, I.S.; Rispens, T.; Muller, F.J.; van Capel, T.M.; Turina, M.C.; Vos, J.B.; Baeten, D.L.; Kapsenberg, M.L.; de Jong, E.C.; et al. Fc gamma receptor-TLR cross-talk elicits pro-inflammatory cytokine production by human M2 macrophages. Nat. Commun. 2014, 5, 5444.
- van der Poel, M.; Hoepel, W.; Hamann, J.; Huitinga, I.; Dunnen, J.d. IgG Immune Complexes Break Immune Tolerance of Human Microglia. J. Immunol. 2020, ji2000130.
- Ben Mkaddem, S.; Hayem, G.; Jönsson, F.; Rossato, E.; Boedec, E.; Boussetta, T.; El Benna, J.; Launay, P.; Goujon, J.-M.; Benhamou, M.; et al. Shifting FcγRIIA-ITAM from activation to inhibitory configuration ameliorates arthritis. J. Clin. Investig. 2014, 124, 3945–3959.
- Breedveld, A.; van Egmond, M. IgA and FcαRI: Pathological Roles and Therapeutic Opportunities. Front. Immunol. 2019, 10, 553.
- Huang, Z.Y.; Barreda, D.R.; Worth, R.G.; Indik, Z.K.; Kim, M.K.; Chien, P.; Schreiber, A.D. Differential kinase requirements in human and mouse Fc-gamma receptor phagocytosis and endocytosis. J. Leukoc. Biol. 2006, 80, 1553–1562.
- Hepburn, A.L.; Mason, J.C.; Wang, S.; Shepherd, C.J.; Florey, O.; Haskard, D.O.; Davies, K.A. Both Fcgamma and complement receptors mediate transfer of immune complexes from erythrocytes to human macrophages under physiological flow conditions in vitro. Clin. Exp. Immunol. 2006, 146, 133–145.
- Hansen, I.S.; Krabbendam, L.; Bernink, J.H.; Loayza-Puch, F.; Hoepel, W.; van Burgsteden, J.A.; Kuijper, E.C.; Buskens, C.J.; Bemelman, W.A.; Zaat, S.A.J.; et al. FcalphaRI co-stimulation converts human intestinal CD103(+) dendritic cells into pro-inflammatory cells through glycolytic reprogramming. Nat. Commun. 2018, 9, 863.
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H.U.; Culemann, S.; Harre, U.; Ackermann, J.A.; Seefried, M.; Kleyer, A.; Uderhardt, S.; et al. Regulation of autoantibody activity by the IL-23-T(H)17 axis determines the onset of autoimmune disease. Nat. Immunol. 2017, 18, 104–113.
- Steffen, U.; Koeleman, C.A.; Sokolova, M.V.; Bang, H.; Kleyer, A.; Rech, J.; Unterweger, H.; Schicht, M.; Garreis, F.; Hahn, J.; et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat. Commun. 2020, 11, 120.
- Gazendam, R.P.; van de Geer, A.; Roos, D.; van den Berg, T.K.; Kuijpers, T.W. How neutrophils kill fungi. Immunol. Rev. 2016, 273, 299–311.
- Li, X.; Leonardi, I.; Semon, A.; Doron, I.; Gao, I.H.; Putzel, G.G.; Kim, Y.; Kabata, H.; Artis, D.; Fiers, W.D.; et al. Response to Fungal Dysbiosis by Gut-Resident CX3CR1(+) Mononuclear Phagocytes Aggravates Allergic Airway Disease. Cell Host Microbe 2018, 24, 847–856.e844.
- Khoyratty, T.E.; Udalova, I.A. Diverse mechanisms of IRF5 action in inflammatory responses. Int J. Biochem. Cell Biol. 2018, 99, 38–42.
- Chistiakov, D.A.; Myasoedova, V.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 2018, 223, 101–111.
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238.
- Weiss, M.; Blazek, K.; Byrne, A.J.; Perocheau, D.P.; Udalova, I.A. IRF5 is a specific marker of inflammatory macrophages in vivo. Mediat. Inflamm. 2013, 2013, 245804.
- Song, S.; De, S.; Nelson, V.; Chopra, S.; LaPan, M.; Kampta, K.; Sun, S.; He, M.; Thompson, C.D.; Li, D.; et al. Inhibition of IRF5 hyper-activation protects from lupus onset and severity. J. Clin. Investig. 2020.
- Yan, J.; Pandey, S.P.; Barnes, B.J.; Turner, J.R.; Abraham, C. T Cell-Intrinsic IRF5 Regulates T Cell Signaling, Migration, and Differentiation and Promotes Intestinal Inflammation. Cell Rep. 2020, 31, 107820.
- Krausgruber, T.; Saliba, D.; Ryzhakov, G.; Lanfrancotti, A.; Blazek, K.; Udalova, I.A. IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 2010, 115, 4421–4430.
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730.
- Ryzhakov, G.; Eames, H.L.; Udalova, I.A. Activation and function of interferon regulatory factor 5. J. Interferon Cytokine Res. 2015, 35, 71–78.
- Hedl, M.; Yan, J.; Abraham, C. IRF5 and IRF5 Disease-Risk Variants Increase Glycolysis and Human M1 Macrophage Polarization by Regulating Proximal Signaling and Akt2 Activation. Cell Rep. 2016, 16, 2442–2455.
- Hedl, M.; Yan, J.; Witt, H.; Abraham, C. IRF5 Is Required for Bacterial Clearance in Human M1-Polarized Macrophages, and IRF5 Immune-Mediated Disease Risk Variants Modulate This Outcome. J. Immunol. 2019, 202, 920–930.
- Balkhi, M.Y.; Fitzgerald, K.A.; Pitha, P.M. Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Mol. Cell Biol. 2008, 28, 7296–7308.
- Corbin, A.L.; Gomez-Vazquez, M.; Berthold, D.L.; Attar, M.; Arnold, I.C.; Powrie, F.M.; Sansom, S.N.; Udalova, I.A. IRF5 guides monocytes toward an inflammatory CD11c+ macrophage phenotype and promotes intestinal inflammation. Sci. Immunol. 2020, 5, eaax6085.
- Heinz, L.X.; Lee, J.; Kapoor, U.; Kartnig, F.; Sedlyarov, V.; Papakostas, K.; César-Razquin, A.; Essletzbichler, P.; Goldmann, U.; Stefanovic, A.; et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7–9. Nature 2020, 581, 316–322.
- Irani, V.; Guy, A.J.; Andrew, D.; Beeson, J.G.; Ramsland, P.A.; Richards, J.S. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol. Immunol. 2015, 67, 171–182.
- Knol, E.F. Requirements for effective IgE cross-linking on mast cells and basophils. Mol. Nutr. Food Res. 2006, 50, 620–624.
- Suurmond, J.; Stoop, J.N.; Rivellese, F.; Bakker, A.M.; Huizinga, T.W.J.; Toes, R.E.M. Activation of human basophils by combined toll-like receptor- and FcεRI-triggering can promote Th2 skewing of naive T helper cells. Eur. J. Immunol. 2014, 44, 386–396.
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcgamma receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108.
- Kubagawa, H.; Oka, S.; Kubagawa, Y.; Torii, I.; Takayama, E.; Kang, D.-W.; Gartland, G.L.; Bertoli, L.F.; Mori, H.; Takatsu, H.; et al. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J. Exp. Med. 2009, 206, 2779–2793.
- Nyamboya, R.A.; Sutton, B.J.; Calvert, R.A. Mapping of the binding site for FcμR in human IgM-Fc. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140266.
- Chen, K.; Magri, G.; Grasset, E.K.; Cerutti, A. Rethinking mucosal antibody responses: IgM, IgG and IgD join IgA. Nat. Rev. Immunol. 2020.
- Shan, M.; Carrillo, J.; Yeste, A.; Gutzeit, C.; Segura-Garzón, D.; Walland, A.C.; Pybus, M.; Grasset, E.K.; Yeiser, J.R.; Matthews, D.B.; et al. Secreted IgD Amplifies Humoral T Helper 2 Cell Responses by Binding Basophils via Galectin-9 and CD44. Immunity 2018, 49, 709–724.
- Hoepel, W.; Golebski, K.; van Drunen, C.M.; den Dunnen, J. Active control of mucosal tolerance and inflammation by human IgA and IgG antibodies. J. Allergy Clin. Immunol. 2020, 146, 273–275.
- den Dunnen, J.; Mes, L.; Hoepel, W.; Smolders, J. Multiple sclerosis: Why we should focus on both sides of the (auto)antibody. Neural Regen Res. 2021, 16, 2422–2424.
- Du Clos, T.W. Pentraxins: Structure, function, and role in inflammation. Isrn Inflamm. 2013, 2013, 379040.
- Agrawal, A.; Singh, P.P.; Bottazzi, B.; Garlanda, C.; Mantovani, A. Pattern recognition by pentraxins. Adv. Exp. Med. Biol. 2009, 653, 98–116.
- Daigo, K.; Inforzato, A.; Barajon, I.; Garlanda, C.; Bottazzi, B.; Meri, S.; Mantovani, A. Pentraxins in the activation and regulation of innate immunity. Immunol. Rev. 2016, 274, 202–217.
- Mold, C.; Du Clos, T.W. C-reactive protein increases cytokine responses to Streptococcus pneumoniae through interactions with Fc gamma receptors. J. Immunol. 2006, 176, 7598–7604.
- Volanakis, J.E.; Kaplan, M.H. Specificity of C-Reactive Protein for Choline Phosphate Residues of Pneumococcal C-Polysaccharide. Proc. Soc. Exp. Biol. Med. 1971, 136, 612–614.
- Bodman-Smith, K.B.; Melendez, A.J.; Campbell, I.; Harrison, P.T.; Allen, J.M.; Raynes, J.G. C-reactive protein-mediated phagocytosis and phospholipase D signalling through the high-affinity receptor for immunoglobulin G (FcgammaRI). Immunology 2002, 107, 252–260.
- Culley, F.J.; Harris, R.A.; Kaye, P.M.; McAdam, K.P.; Raynes, J.G. C-reactive protein binds to a novel ligand on Leishmania donovani and increases uptake into human macrophages. J. Immunol. 1996, 156, 4691–4696.
- Peisajovich, A.; Marnell, L.; Mold, C.; Du Clos, T.W. C-reactive protein at the interface between innate immunity and inflammation. Expert Rev. Clin. Immunol. 2008, 4, 379–390.
- Rhodes, B.; Fürnrohr, B.G.; Vyse, T.J. C-reactive protein in rheumatology: Biology and genetics. Nat. Rev. Rheumatol. 2011, 7, 282–289.
- Hart, S.P.; Alexander, K.M.; MacCall, S.M.; Dransfield, I. C-reactive protein does not opsonize early apoptotic human neutrophils, but binds only membrane-permeable late apoptotic cells and has no effect on their phagocytosis by macrophages. J. Inflamm. 2005, 2, 5.
- Volanakis, J.E.; Wirtz, K.W. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature 1979, 281, 155–157.
- Li, Y.P.; Mold, C.; Du Clos, T.W. Sublytic complement attack exposes C-reactive protein binding sites on cell membranes. J. Immunol. 1994, 152, 2995–3005.
- Du Clos, T.W.; Marnell, L.; Zlock, L.R.; Burlingame, R.W. Analysis of the binding of C-reactive protein to chromatin subunits. J. Immunol. 1991, 146, 1220–1225.
- Du Clos, T.W.; Zlock, L.T.; Rubin, R.L. Analysis of the binding of C-reactive protein to histones and chromatin. J. Immunol. 1988, 141, 4266–4270.
- Newling, M.; Sritharan, L.; van der Ham, A.J.; Hoepel, W.; Fiechter, R.H.; de Boer, L.; Zaat, S.A.J.; Bisoendial, R.J.; Baeten, D.L.P.; Everts, B.; et al. C-Reactive Protein Promotes Inflammation through FcgammaR-Induced Glycolytic Reprogramming of Human Macrophages. J. Immunol. 2019, 203, 225–235.
- Hoepel, W.; Allahverdiyeva, S.; Harbiye, H.; de Taeye, S.W.; van der Ham, A.J.; de Boer, L.; Zaat, S.A.J.; van Weeghel, M.; Baeten, D.L.P.; Houtkooper, R.H.; et al. IgG Subclasses Shape Cytokine Responses by Human Myeloid Immune Cells through Differential Metabolic Reprogramming. J. Immunol. 2020, 205, 3400–3407.
- Diskin, C.; Pålsson-McDermott, E.M. Metabolic Modulation in Macrophage Effector Function. Front. Immunol. 2018, 9, 270.
- Izquierdo, E.; Cuevas, V.D.; Fernández-Arroyo, S.; Riera-Borrull, M.; Orta-Zavalza, E.; Joven, J.; Rial, E.; Corbi, A.L.; Escribese, M.M. Reshaping of Human Macrophage Polarization through Modulation of Glucose Catabolic Pathways. J. Immunol. 2015, 195, 2442–2451.
- Wei, J.; Tang, D.; Lu, C.; Yang, J.; Lu, Y.; Wang, Y.; Jia, L.; Wang, J.; Ru, W.; Lu, Y.; et al. Irf5 deficiency in myeloid cells prevents necrotizing enterocolitis by inhibiting M1 macrophage polarization. Mucosal Immunol. 2019, 12, 888–896.
- Hoepel, W.; Chen, H.-J.; Allahverdiyeva, S.; Manz, X.; Aman, J.; Bonta, P.; Brouwer, P.; de Taeye, S.; Caniels, T.; van der Straten, K.; et al. Anti-SARS-CoV-2 IgG from severely ill COVID-19 patients promotes macrophage hyper-inflammatory responses. Sci. Transl. Med. 2021, eabf8654.
- Castro-Dopico, T.; Dennison, T.W.; Ferdinand, J.R.; Mathews, R.J.; Fleming, A.; Clift, D.; Stewart, B.J.; Jing, C.; Strongili, K.; Labzin, L.I.; et al. Anti-commensal IgG Drives Intestinal Inflammation and Type 17 Immunity in Ulcerative Colitis. Immunity 2019, 50, 1099–1114.e1010.
- Newling, M.; Fiechter, R.H.; Sritharan, L.; Hoepel, W.; van Burgsteden, J.A.; Hak, A.E.; van Vollenhoven, R.F.; van de Sande, M.G.H.; Baeten, D.L.P.; den Dunnen, J. Dysregulated Fcγ receptor IIa-induced cytokine production in dendritic cells of lupus nephritis patients. Clin. Exp. Immunol. 2020, 199, 39–49.
- Gandhi, R.T.; Lynch, J.B.; del Rio, C. Mild or Moderate Covid-19. N. Engl. J. Med. 2020, 383, 1757–1766.
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374.
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362.
- Larsen, M.D.; de Graaf, E.L.; Sonneveld, M.E.; Plomp, H.R.; Nouta, J.; Hoepel, W.; Chen, H.-J.; Linty, F.; Visser, R.; Brinkhaus, M.; et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 2021, 371, eabc8378.
- Dekkers, G.; Treffers, L.; Plomp, R.; Bentlage, A.E.H.; de Boer, M.; Koeleman, C.A.M.; Lissenberg-Thunnissen, S.N.; Visser, R.; Brouwer, M.; Mok, J.Y.; et al. Decoding the Human Immunoglobulin G-Glycan Repertoire Reveals a Spectrum of Fc-Receptor- and Complement-Mediated-Effector Activities. Front. Immunol. 2017, 8, 877.
- Bye, A.P.; Hoepel, W.; Mitchell, J.L.; Jégouic, S.; Loureiro, S.; Sage, T.; de Taeye, S.; van Gils, M.; Kriek, N.; Cooper, N.; et al. Aberrant glycosylation of anti-SARS-CoV-2 IgG is a pro-thrombotic stimulus for platelets. bioRxiv 2021.
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4, e123158.
- Su, Z.; Xie, Q.; Wang, Y.; Li, Y. Abberant Immunoglobulin G Glycosylation in Rheumatoid Arthritis by LTQ-ESI-MS. Int. J. Mol. Sci. 2020, 21, 2045.
- Harre, U.; Lang, S.C.; Pfeifle, R.; Rombouts, Y.; Frühbeißer, S.; Amara, K.; Bang, H.; Lux, A.; Koeleman, C.A.; Baum, W.; et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat. Commun. 2015, 6, 6651.
- Wuhrer, M.; Selman, M.H.J.; McDonnell, L.A.; Kümpfel, T.; Derfuss, T.; Khademi, M.; Olsson, T.; Hohlfeld, R.; Meinl, E.; Krumbholz, M. Pro-inflammatory pattern of IgG1 Fc glycosylation in multiple sclerosis cerebrospinal fluid. J. Neuroinflamm. 2015, 12, 235.
- Toong, C.; Adelstein, S.; Phan, T.G. Clearing the complexity: Immune complexes and their treatment in lupus nephritis. Int. J. Nephrol. Renovasc. Dis. 2011, 4, 17–28.
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196.
- Klareskog, L.; Ronnelid, J.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 2008, 26, 651–675.
- El Bannoudi, H.; Ioan-Facsinay, A.; Toes, R.E. Bridging autoantibodies and arthritis: The role of fc receptors. Curr. Top. Microbiol. Immunol. 2014, 382, 303–319.
- Means, T.K.; Latz, E.; Hayashi, F.; Murali, M.R.; Golenbock, D.T.; Luster, A.D. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Investig. 2005, 115, 407–417.
- Devarapu, S.K.; Anders, H.-J. Toll-like receptors in lupus nephritis. J. Biomed. Sci. 2018, 25, 35.
- Kim, J.M.; Park, S.H.; Kim, H.Y.; Kwok, S.K. A Plasmacytoid Dendritic Cells-Type I Interferon Axis Is Critically Implicated in the Pathogenesis of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2015, 16, 14158–14170.
- Kim, W.-U.; Sreih, A.; Bucala, R. Toll-like receptors in systemic lupus erythematosus; prospects for therapeutic intervention. Autoimmun. Rev. 2009, 8, 204–208.
- Weber, M.S.; Hemmer, B.; Cepok, S. The role of antibodies in multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 239–245.
- Mavragani, C.P.; Tzioufas, A.G.; Moutsopoulos, H.M. Sjögren’s syndrome: Autoantibodies to cellular antigens. Clinical and molecular aspects. Int. Arch. Allergy Immunol. 2000, 123, 46–57.
- Pan, M.; Liu, X.; Zheng, J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin. Exp. Dermatol. 2011, 36, 703–707.
- Melief, J.; Koning, N.; Schuurman, K.G.; Van De Garde, M.D.; Smolders, J.; Hoek, R.M.; Van Eijk, M.; Hamann, J.; Huitinga, I. Phenotyping primary human microglia: Tight regulation of LPS responsiveness. Glia 2012, 60, 1506–1517.
- Kramer, J.M. Early events in Sjögren’s Syndrome pathogenesis: The importance of innate immunity in disease initiation. Cytokine 2014, 67, 92–101.
- Mehra, S.; Walker, J.; Patterson, K.; Fritzler, M.J. Autoantibodies in systemic sclerosis. Autoimmun. Rev. 2013, 12, 340–354.
- Tsokos, G.C. Systemic Lupus Erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121.
- Ban, T.; Sato, G.R.; Tamura, T. Regulation and role of the transcription factor IRF5 in innate immune responses and systemic lupus erythematosus. Int. Immunol. 2018, 30, 529–536.
- Brito-Zerón, P.; Baldini, C.; Bootsma, H.; Bowman, S.J.; Jonsson, R.; Mariette, X.; Sivils, K.; Theander, E.; Tzioufas, A.; Ramos-Casals, M. Sjögren syndrome. Nat. Rev. Dis. Primers 2016, 2, 16047.
- Tang, L.; Chen, B.; Ma, B.; Nie, S. Association between IRF5 polymorphisms and autoimmune diseases: A meta-analysis. Genet. Mol. Res. 2014, 13, 4473–4485.
- Rochereau, N.; Roblin, X.; Michaud, E.; Gayet, R.; Chanut, B.; Jospin, F.; Corthésy, B.; Paul, S. NOD2 deficiency increases retrograde transport of secretory IgA complexes in Crohn’s disease. Nat. Commun. 2021, 12, 261.
- Chang, M.K.; Binder, C.J.; Torzewski, M.; Witztum, J.L. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2002, 99, 13043–13048.
- Pilely, K.; Fumagalli, S.; Rosbjerg, A.; Genster, N.; Skjoedt, M.O.; Perego, C.; Ferrante, A.M.R.; De Simoni, M.G.; Garred, P. C-Reactive Protein Binds to Cholesterol Crystals and Co-Localizes with the Terminal Complement Complex in Human Atherosclerotic Plaques. Front. Immunol. 2017, 8, 1040.
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-Reactive Protein and Low-Density Lipoprotein Cholesterol Levels in the Prediction of First Cardiovascular Events. N. Engl. J. Med. 2002, 347, 1557–1565.
- Oliviero, F.; Lo Nigro, A.; Bernardi, D.; Giunco, S.; Baldo, G.; Scanu, A.; Sfriso, P.; Ramonda, R.; Plebani, M.; Punzi, L. A comparative study of serum and synovial fluid lipoprotein levels in patients with various arthritides. Clin. Chim. Acta 2012, 413, 303–307.

