Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tyler Beck | + 1366 word(s) | 1366 | 2021-05-25 11:26:23 | | | |
| 2 | Vicky Zhou | Meta information modification | 1366 | 2021-06-03 03:13:55 | | | | |
| 3 | Vicky Zhou | Meta information modification | 1366 | 2021-06-08 09:00:24 | | | | |
| 4 | Vicky Zhou | Meta information modification | 1366 | 2021-06-08 09:01:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Beck, T. Cytochrome Inhibitors. Encyclopedia. Available online: https://encyclopedia.pub/entry/10445 (accessed on 28 June 2026).
Beck T. Cytochrome Inhibitors. Encyclopedia. Available at: https://encyclopedia.pub/entry/10445. Accessed June 28, 2026.
Beck, Tyler. "Cytochrome Inhibitors" Encyclopedia, https://encyclopedia.pub/entry/10445 (accessed June 28, 2026).
Beck, T. (2021, June 02). Cytochrome Inhibitors. In Encyclopedia. https://encyclopedia.pub/entry/10445
Beck, Tyler. "Cytochrome Inhibitors." Encyclopedia. Web. 02 June, 2021.
Copy Citation
Major cytochrome inhibitors may result in drug–drug interactions.
CYP3A4
CYP2D6
CYP2C19
CYP2C9
CYP1A2
cheminformatics
cytochrome
1. Introduction
Roughly seventy-four percent (74%) of all physician office visits involve drug therapy, with 2.9 billion drugs prescribed annually [1]. These prescriptions accounted for USD 335 billion in patient costs in 2020 [2]. Half of Americans take at least one prescription drug and nearly a quarter of persons use three or more medications concomitantly [1]. An estimated 6.7% of hospitalized patients experience an adverse drug reaction with a fatality rate of 0.32%, responsible for 106,000 deaths annually [3]. These data suggest adverse drug reactions are the fourth leading cause of death in the United States [3][4][5]. Furthermore, these statistics exclude ambulatory adverse drug reactions and are therefore likely an underrepresentation of the morbidity and mortality burden of adverse drug reactions. Nearly 350,000 adverse drug reactions occur in nursing homes annually, where polypharmacy is commonplace [6]. Many of these drug–drug interactions are avoidable with proper patient medication management and by deprescribing. While efforts to reduce drug–drug interactions are important, a reduction in the number of medications cannot always be achieved without impacting therapeutic outcomes.
Adverse drug reactions commonly occur due to drug–drug interactions as a result of major cytochrome P450 (CYP) inhibition [7]. CYPs are heme-containing membrane-bound enzymes that predominantly reside in the smooth endoplasmic reticulum and mitochondria of hepatocytes and in the intestines [8][9]. In mammals, 57 CYP isoforms have been identified with the function of performing the oxidative metabolism of xenobiotics and endogenous compounds [8]. Of the 57 CYP isoforms, 5 CYPs (CYPs 3A4, 2D6, 2C19, 2C9, and 1A2) are responsible for the metabolism of more than 80% of clinically used drugs [7] (Figure 1). Enzyme inhibition occurs when two co-administered drugs share an identical mechanism of biotransformation and compete for metabolism in the same enzyme receptor site [9]. As such, the more potent inhibitor will prevail, interrupting biotranformation, resulting in the diminished metabolism of the competing drug. This results in elevated serum drug levels of the unmetabolized drug, increasing the probability of adverse toxicological outcomes. This is especially important for drugs with a narrow therapeutic index, such as chemotherapeutics. It is important to note that inhibitors of major cytochromes can promote adverse drug reactions when administered as monotherapy. Inhibitors of major cytochromes may alter their own metabolism at high doses, leading to toxic plasma drug accumulation. These interactions pose a significant safety risk and should be examined closer during the initial drug design process. The CYP system and drug biotransformation has been reviewed extensively elsewhere [10].
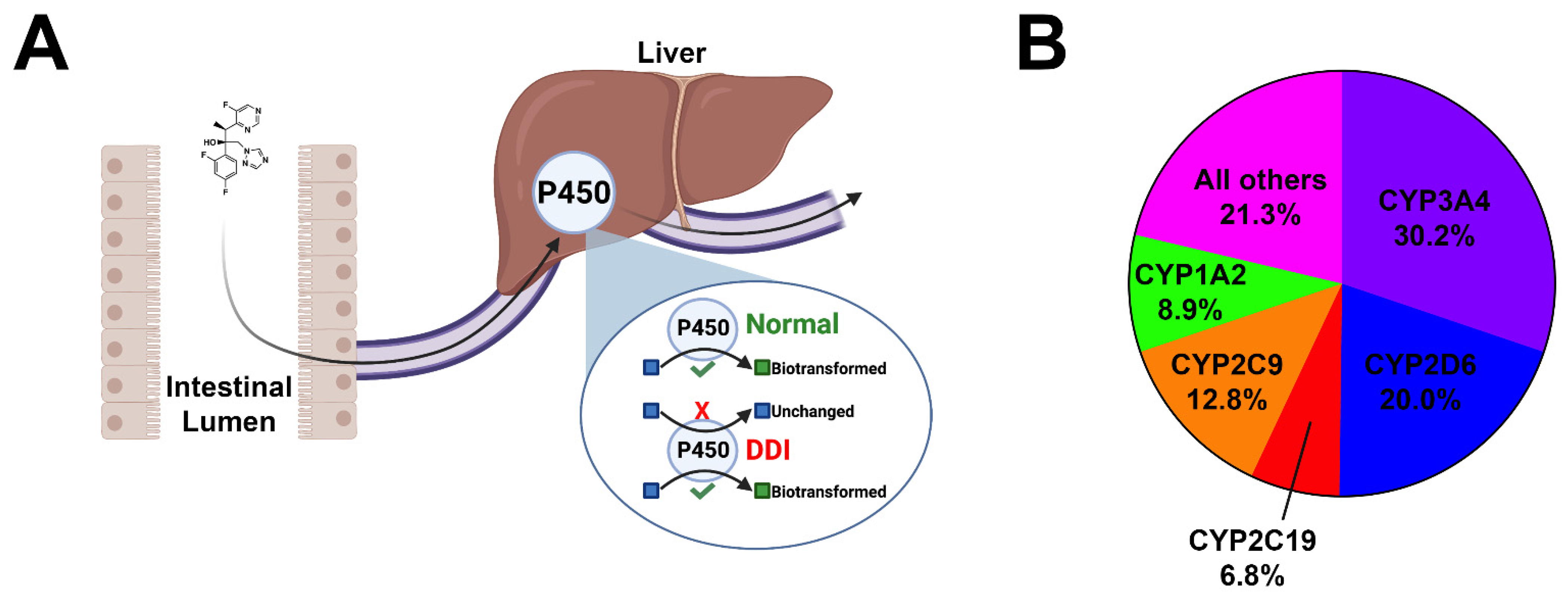 Figure 1. Cartoon depiction of cytochrome inhibition and pie chart. (A) Orally bioavailable drugs are absorbed from the intestinal lumen into the mesenteric capillaries and transported to the liver via the portal vein. Many drugs undergo phase I biotransformation by major cytochromes within hepatocytes. This process enzymatically converts lipid-soluble compounds to more water-soluble compounds to facilitate the excretion. In the event of polypharmacy and/or the co-administration of CYP inhibitors, serum accumulation of unmetabolized drugs may occur, leading to untoward toxicities. (B) Pie chart demonstrating the breakdown of the five major cytochromes and their contributions to the oxidative metabolism of xenobiotics and organic endogenous molecules.
Figure 1. Cartoon depiction of cytochrome inhibition and pie chart. (A) Orally bioavailable drugs are absorbed from the intestinal lumen into the mesenteric capillaries and transported to the liver via the portal vein. Many drugs undergo phase I biotransformation by major cytochromes within hepatocytes. This process enzymatically converts lipid-soluble compounds to more water-soluble compounds to facilitate the excretion. In the event of polypharmacy and/or the co-administration of CYP inhibitors, serum accumulation of unmetabolized drugs may occur, leading to untoward toxicities. (B) Pie chart demonstrating the breakdown of the five major cytochromes and their contributions to the oxidative metabolism of xenobiotics and organic endogenous molecules.Traditionally, pharmacokinetics has been neglected in preclinical drug design. Roughly four in five investigational new drugs fail as a result of absorption, distribution, metabolism, excretion and toxicity (ADMET) issues [11]. Tools that assist in ADMET prediction may be advantageous in early drug design by reducing the drug attrition rate in advanced preclinical- and early clinical-stage studies. In particular, the design of drugs devoid of major cytochrome inhibition would reduce the burden of drug–drug interactions in patients where polypharmacy is unavoidable. Several reviews exist that focus on platforms that utilize a diverse range of prediction methods [12][13][14][15][16][17][18][19][20]. Many of these reviews focus on outdated tools, as well as platforms that are exclusively available through a fee-based licensing model. More recently, numerous machine learning-based platforms have emerged that are user-friendly, are not as computationally expensive as pre-existing methods, and demonstrate improved prediction accuracy.
2. Machine Learning-Based Methods
Current in vitro and in vivo experimental approaches to investigate drug ADMET properties are highly effective in assessing pharmacokinetics; however, such approaches are extraordinarily expensive and are commonly performed late in drug development during IND-enabling studies [13]. In silico investigation of CYP interactions early in drug development allows investigators to select for compounds that are less likely to fail in late-stage preclinical or early clinical trials due to undesirable pharmacokinetics (Figure 2). Such tools exist and are effective at predicting CYP inhibitors, common sites of metabolism (SoMs), and prospective drug metabolites. Hereby we highlight computational approaches available for ADMET predictions, focusing primarily on CYP inhibition and metabolism. The aim of this discussion will be to highlight emerging platforms that utilize machine learning-based methods, with a special emphasis placed on applications that are free to the user and available through a public webserver. A complete overview of platforms available through a fee-based licensing model, as well as older free prediction methods and methods for analyzing CYP molecular dynamics, are reviewed elsewhere [13][14][15][16][17][18][19][20].
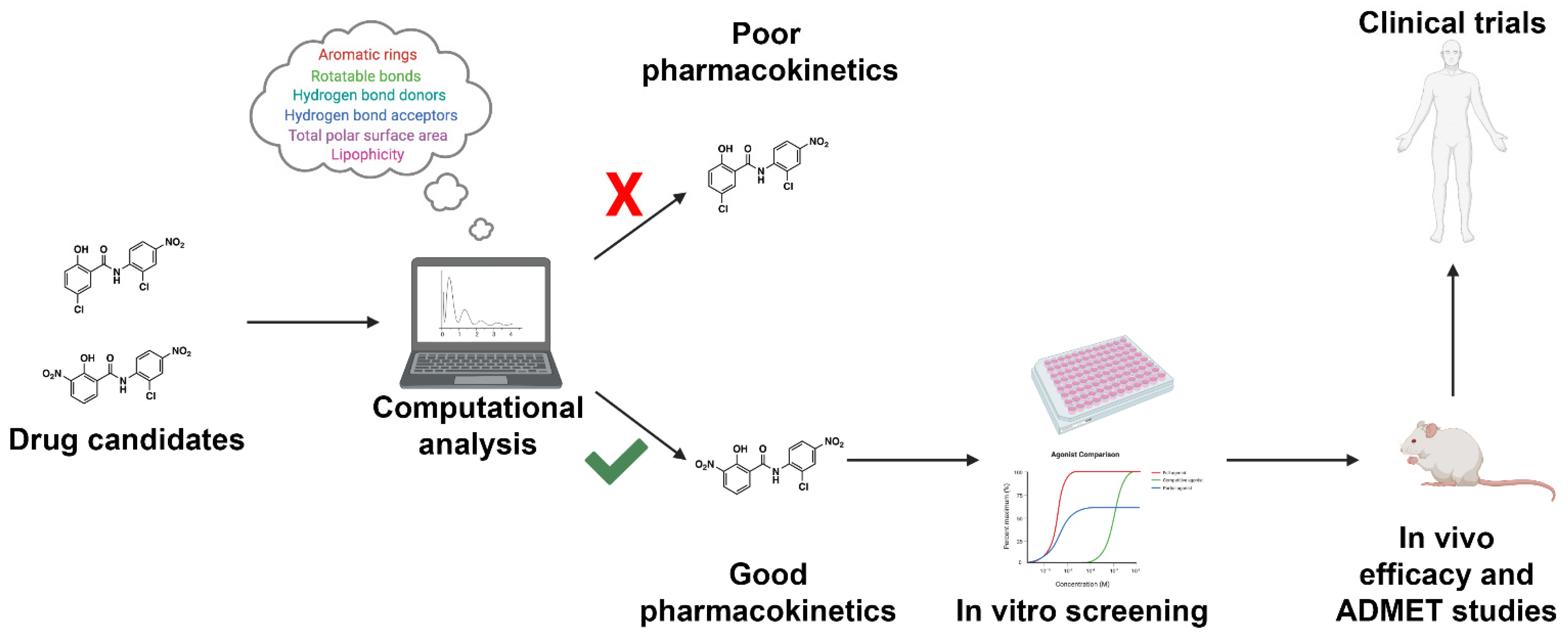 Figure 2. Workflow for incorporating ADMET predictions into early-stage drug design and development. Drug candidates can be readily screened using high-throughput computational approaches. Drugs with acceptable pharmacokinetic parameters as defined by the investigator will proceed to in vitro screening. Drugs with unacceptable pharmacokinetic properties will be triaged early in drug development. The following approach will result in a reduction in ADMET-related drug failures, reducing the cost of drug discovery efforts.
Figure 2. Workflow for incorporating ADMET predictions into early-stage drug design and development. Drug candidates can be readily screened using high-throughput computational approaches. Drugs with acceptable pharmacokinetic parameters as defined by the investigator will proceed to in vitro screening. Drugs with unacceptable pharmacokinetic properties will be triaged early in drug development. The following approach will result in a reduction in ADMET-related drug failures, reducing the cost of drug discovery efforts.Various platforms capable of making enzyme-inhibitor predictions exist. These techniques use an array of methods, ranging from molecular docking with flexible multi-dimensional QSAR to machine learning based approaches. It should be noted that flexible docking-based methods, such as VirtualToxLab, are computationally expensive, take longer to receive results, and commonly require a software license [13][21]. Machine learning-based tools are capable of producing accurate results in a timely fashion and can be hosted via a publicly available web server. These platforms tend to be more user-friendly, and are commonly able to screen a large number of structures. These platforms each use unique descriptors and utilize their own methods of data generation. As such, the accuracy of said platforms is subject to variability, contingent on the descriptors and molecular fingerprints used, the number of input structures used to build and train the model, and how the data are interpreted. In this section, we review the best publicly available machine learning-based platforms free to the scientific community.
3. Conclusions
More than half of drug failures are a result of poor ADMET. More specifically, drug–drug interactions as a result of major cytochrome inhibition are commonplace and can lead to life-threatening adverse drug reactions. A majority of pre-clinical-stage drug discovery programs focus solely on binding affinity and selectivity [22]. Although binding affinity is of utmost importance, ADMET properties ensure that a drug is properly absorbed, distributed, and cleared; features that equally contribute to the success of a drug in the clinic. As such, in silico approaches have been developed to be introduced during early drug screening, with the goal of reducing the rate of drug attrition (Table 1).
Table 1. Summary of publicly available machine learning-based methods for cytochrome inhibition and metabolism.
| Name | CYPs | Prediction Type | ML Method | No. Structures | Avg. Accuracy 1 | Additional Features |
|---|---|---|---|---|---|---|
| pkCSM | 3A4, 2D6, 2C19, 2C9, 1A2 |
Inhibition | Graph-based signatures |
18,000 | 0.810 (0.780–0.853) |
Comprehensive ADMET predictions (23 total) |
| DeepCYP | 3A4, 2D6, 2C19, 2C9, 1A2 |
Inhibition | Multitask autoencoder deep neural network | 13,000 | 0.864 (0.809–0.968) |
Assigns probabilities for CYP inhibition |
| SuperCYPs-Pred | 3A4, 2D6, 2C19, 2C9, 1A2 |
Inhibition | Random forests | 41,963 2 | 0.930 (0.840–0.970) |
Assigns probabilities for CYP inhibition |
| vNN-ADMET | 3A4, 2D6, 2C19, 2C9, 1A2 |
Inhibition | Variable nearest neighbors | 6261 | 0.890 (0.870–0.910) |
|
| AdmetSAR 2.0 | 3A4, 2D6, 2C19, 2C9, 1A2 |
Inhibition | Random forests, support vector machines, k-nearest neighbors |
96,000 3 | 0.784 (0.645–0.855) |
Comprehensive ADMET predictions (47 total); ADMETopt for lead optimization |
| SwissADME | 3A4, 2D6l 2C19, 2C9, 1A2 |
Inhibition | Support vector machines | 16,561 4 | 0.794 (0.720–0.800) |
Predictions of physicochemical properties, pharmacokinetics, and drug likeness; high throughput |
| CypRules | 3A4, 2D6, 2C19, 2C9, 1A2 |
Inhibition | Decision trees | 16,561 | 0.812 (0.730–0.900) |
High throughput |
| CypReact | 3A4, 2E1, 2D6,2C19, 2C9, 2C8, 2B6, 2A6, 1A2 |
Sites of metabolism |
LBM learning algorithm |
2685 | Unavailable | Metabolite predictions; Additional CYPs |
1 Cross-validation accuracy. 2 Sum of the number of compounds used for all five major cytochromes. The dataset includes inhibitors and non-inhibitors and may include duplicates. 3 Includes compounds used to build ADMET models in addition to CYP inhibition models. 4 Includes compounds used to establish physicochemical properties, pharmacokinetics, drug likeness and medicinal chemistry friendliness models.
References
- Therapeutic Drug Use. Available online: (accessed on 28 February 2021).
- Mikulic, M. Prescription Drug Expenditure in the United States 1960–2020. Available online: (accessed on 28 January 2021).
- Donaldson, M.S.; Corrigan, J.M.; Kohn, L.T. (Eds.) In to Err Is Human: Building a Safer Health System; National Academies Press: Washington, DC, USA, 2000.
- Lazarou, J.; Pomeranz, B.H.; Corey, P.N. Incidence of Adverse Drug Reactions in Hospitalized Patients. JAMA 1998, 279, 1200–1205.
- Preventable Adverse Drug Reactions: A Focus on Drug Interactions. Available online: (accessed on 2 February 2021).
- Gurwitz, J.H.; Field, T.S.; Avorn, J.; McCormick, D.; Jain, S.; Eckler, M.; Benser, M.; Edmondson, A.C.; Bates, D.W. Incidence and preventability of adverse drug events in nursing homes. Am. J. Med. 2000, 109, 87–94.
- Banerjee, P.; Dunkel, M.; Kemmler, E.; Preissner, R. SuperCYPsPred—A web server for the prediction of cytochrome activity. Nucleic Acids Res. 2020, 48, W580–W585.
- Redlich, G.; Zanger, U.M.; Riedmaier, S.; Bache, N.; Giessing, A.B.M.; Eisenacher, M.; Stephan, C.; Meyer, H.E.; Jensen, O.N.; Marcus, K. Distinction between Human Cytochrome P450 (CYP) Isoforms and Identification of New Phosphorylation Sites by Mass Spectrometry. J. Proteome Res. 2008, 7, 4678–4688.
- Ogu, C.C.; Maxa, J.L. Drug Interactions Due to Cytochrome P450. In Baylor University Medical Center Proceedings; Informa UK Limited: London, UK, 2000; Volume 13, pp. 421–423.
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery. In A Comprehensive Guide to Toxicology in Preclinical Drug Development; Faqi, A.S., Ed.; Academic Press: Cambridge, MA, USA, 2013.
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pract. Oncol. 2013, 4, 263–268.
- Raunio, H.; Kuusisto, M.; Juvonen, R.O.; Pentikainen, O.T. Modeling of interactions between xenobiotics and cytochrome P450 (CYP) enzymes. Front. Pharmacol. 2015, 6, 123.
- Tyzack, J.D.; Kirchmair, J. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des. 2019, 93, 377–386.
- Martiny, V.Y.; Carbonell, P.; Chevillard, F.; Moroy, G.; Nicot, A.B.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Integrated structure- and ligand-basedin silicoapproach to predict inhibition of cytochrome P450 2D6. Bioinformatics 2015, 31, 3930–3937.
- Refsgaard, H.H.F.; Jensen, B.F.; Christensen, I.T.; Hagen, N.; Brockhoff, P.B. In silico prediction of cytochrome P450 inhibitors. Drug Dev. Res. 2006, 67, 417–429.
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. MetaPASS: A Web Application for Analyzing the Biological Activity Spectrum of Organic Compounds Taking into Account their Biotransformation. Mol. Inform. 2021, 40, 2000231.
- Kar, S. Recent Advances of Computational Modeling for Predicting Drug Metabolism: A Perspective. Curr. Drug Metab. 2018, 18, 1106–1122.
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404.
- Shaik, S.; Chen, H.; Usharani, D.; Thiel, W. QM/MM Studies of Structure and Reactivity of Cytochrome P450 Enzymes: Methodology and Selected Applications. Early Drug Dev. 2014, 311, 133–178.
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54.
- Vedani, A.; Dobler, M.; Smieško, M. VirtualToxLab—A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 2012, 261, 142–153.
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
918
Revisions:
4 times
(View History)
Update Date:
08 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No