Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dennis Timmerman | + 2547 word(s) | 2547 | 2021-05-28 05:45:28 | | | |
| 2 | Peter Tang | Meta information modification | 2547 | 2021-06-03 08:15:23 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Timmerman, D. TP53 Pathway in Embryonic/Somatic Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/10425 (accessed on 24 July 2026).
Timmerman D. TP53 Pathway in Embryonic/Somatic Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/10425. Accessed July 24, 2026.
Timmerman, Dennis. "TP53 Pathway in Embryonic/Somatic Cells" Encyclopedia, https://encyclopedia.pub/entry/10425 (accessed July 24, 2026).
Timmerman, D. (2021, June 02). TP53 Pathway in Embryonic/Somatic Cells. In Encyclopedia. https://encyclopedia.pub/entry/10425
Timmerman, Dennis. "TP53 Pathway in Embryonic/Somatic Cells." Encyclopedia. Web. 02 June, 2021.
Copy Citation
The P53 pathway is the most important cellular pathway to maintain genomic and cellular integrity, both in embryonic and non-embryonic cells. Stress signals induce its activation, initiating autophagy or cell cycle arrest to enable DNA repair.
P53 pathway
cancers
mutations
embryonic and somatic cells
1. Embryonic Stem Cells and Germ Cell Tumors Versus Somatic Cells
The development of multicellular, sexually reproductive organisms starts with the fusion of a spermatozoa and an oocyte that created a diploid zygote (depicted in Figure 1). Then, the zygote divides, forming a cluster of undifferentiated cells (i.e., blastomeres) known as the morula. The first step of lineage differentiation occurs during the subsequent blastocyst stage in which the embryoblast (i.e., the inner cell mass) and the trophectoderm are formed [1][2]. The trophectoderm develops into all extra-embryonic structures, whereas the embryoblast, which consists of pluripotent embryonic stem (ES) cells, develops into all embryonic structures [2]. ES cells are transiently present, can self-renew and give rise to all three embryonic germ layers during gastrulation (i.e., the ecto-, meso-, and endoderm), which ultimately will give rise to all cell lineages of the adult organism [2]. Additionally, ES cells give rise to the primordial germ cells (PGC) in the embryonic yolk sac, which subsequently migrate toward developing gonads to give rise to the germ line [2][3] (see Figure 1).
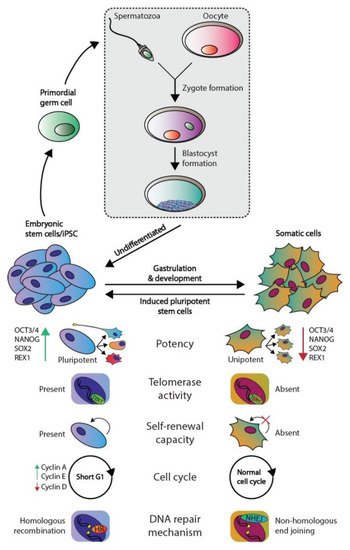
Figure 1. Phenotypic differences between embryonic and somatic cells. Fertilization of an oocyte by a spermatozoon forms a zygote, which subsequently develops the blastocyst. The blastocyst contains embryonic stem (ES) cells that eventually give rise to all somatic cell types. ES and somatic cells can be distinguished according to multiple aspects: their potency, telomerase activity, self-renewal capacity, cell cycle regulation, and preferred DNA repair mechanism. Both germ cells and germ cells tumors are derived from ES derived PGCs and therefore harbor the same embryonal characteristics.
The pluripotent nature of ES cells is characterized by the expression of pluripotency markers including but not limited to OCT4, NANOG, SOX2, and REX1 (Figure 1) [4]. In contrast, somatic cells are differentiated and thus lineage-restricted and unipotent [5]. Therefore, these cells seldomly give rise to other progeny than the identity of the cells themselves [5]. In addition, due to their capacity to self-renew, ES cells can be cultured indefinitely in vitro while retaining their embryonic state as well as a stable genome. This immortality is facilitated by active telomerase, which is an enzyme that extends telomeric repeats that are otherwise lost due to the end-replication problem after each cell cycle (approximately 50–150 base pairs are lost per cycle) [6][7] (Figure 1). Conversely, somatic cells have a restricted lifespan due to the lack of active telomerase and induce cellular senescence when a critical telomere length is reached [6][7]. Furthermore, ES cells also regulate the cell cycle differently as these lack Cyclin D expression yet continuously express Cyclin A and E, which leads to a significantly shortened G1-phase when compared to somatic cells [8]. This characteristic also facilitates the tendency for ES cells to initiate apoptosis rather than cell cycle arrest and DNA repair when DNA damage occurs, the latter of which occurs predominantly in the G1 phase after cell cycle arrest [9][10]. The preference for apoptosis in these early embryonic cells is considered a failsafe mechanism to preserve the genetic integrity of their multitudinous progeny. In rare occasions, ES cells can initiate DNA repair; however, these cells then opt for error-free homologous recombination (HR) to repair double-strand breaks, whereas somatic cells predominantly employ error-prone non-homologous end joining (NHEJ) [9]. Finally, a zygote is known to lose its inherited DNA methylation pattern immediately after fertilization and subsequent changes in DNA methylation occur during early embryonic development as various cell lineages arise. Thus, DNA methylation patterns between ES and somatic cells vary significantly [11][12].
Of note, a discovery made over a decade ago demonstrated that differentiated somatic human cells (e.g., adult human fibroblasts) could be reprogrammed to a pluripotent state reminiscent of ES cells (i.e., nuclear reprogramming) [13]. These cells are known as induced pluripotent stem cells (iPSCs) and, in theory, they can also be maintained in a pluripotent state indefinitely. iPSCs are originally generated in vitro by overexpressing a cocktail of four essential genes (OCT3/4, c-MYC, SOX2, and KLF4) in the differentiated cells [14].
Moreover, many of the characteristics of ES cells are strongly conserved in germ cell tumors (GCTs), which represent a heterogeneous cluster of solid tumors that originate from both ES cells and PGCs [3]. Of note, the parallels observed between ES cells and GCTs do not apply to the teratomas, which is a GCT subtype that consists of fully differentiated cells and harbor significant differences in their (epi)genetic makeup and clinical response (i.e., inherent chemotherapeutic resistance) compared to other GCT subtypes [3][15][16][17]. An example of the similarities observed between ES cells and GCTs is that the latter also express multiple pluripotency markers including OCT3/4 and NANOG and have active telomerase activity, again with the exception of teratoma [3]. Moreover, it is widely accepted that in response to DNA damage, GCTs also favor apoptosis, which is thought to underlie their unique sensitivity to platinum-based chemotherapeutics, including cisplatin (a key component of GCT treatment) [9][10][18]. This is supported by additional evidence demonstrating that GCTs have low expression of genes involved in cell cycle arrest and a deregulated G1-S phase checkpoint, thus potentially also preventing the activation of DNA repair pathways [18][19][20][21][22][23]. Moreover, the apoptotic response of ES cells and GCTs in response to DNA damage has been attributed to the well-known P53 pathway; however, at least in GCTs, it remains a topic of debate [18][21][24][25][26][27][28][29][30][31][32][33][34][35][36][37]. The P53 pathway has been dubbed the guardian of the genome due to its integral role in maintaining genomic integrity among all cell types. This pathway acts in response to cellular stress and lies at the apex of a plethora of downstream signaling pathways including cell cycle arrest, DNA repair, and apoptosis. The deregulation of this pathway has been observed in cancer and is strongly associated to many aspects of tumorigenesis. For instance, GCTs most often express high levels of wild-type (WT) TP53, and it has been suggested that the P53 pathway is (partially) responsible for their characteristic to enter apoptosis in response to platinum-based chemotherapeutics (e.g., cisplatin) which is reminiscent of ES cells in response to DNA damage [10][18][21][23][25][26][27][28][29][33][34][35][36][37]. In addition, it has been suggested that the P53 pathway is deregulated through multiple mechanisms in GCTs that have acquired resistance to chemotherapy, including cisplatin [15][18][38][39][40][41][42].
2. Protecting Genomic Integrity
Genomic changes during development serve as the driving force of evolution; however, these may also negatively impact multiple cell lineages with possible detrimental effects on both the individual and their offspring. Therefore, protection of the genomic integrity is paramount for a species to survive and commences during early embryonic development [23]. For example, the oocyte expresses and deposits multiple mRNAs that protect the zygote against DNA damage during the first cellular divisions [43]. As mentioned before, there have been several indications of a robust protective mechanism in ES cells including the removal of mutated ES cells from the stem cell pool through apoptosis, which is a characteristic that is seemingly conserved in GCTs [7][12][44]. Alternatively, ES cells may also be instructed to differentiate through transcriptional inhibition of the pluripotency factor NANOG, which also effectively removes ES cells from the stem cell pool [45]. These mechanisms in combination with lacking a G1 checkpoint are thought to result in a 100-fold lower frequency of accumulating mutations in at least murine ES cells compared to mouse embryonic fibroblasts, which resemble the somatic cells [9][23]. Conversely to ES cells and GCTs, somatic cells utilize different pathways (DNA repair rather than apoptosis) to protect their genome that enable these to survive while risking an increased mutational load [9][23].
3. P53 Pathway
As mentioned before, the P53 pathway is essential in maintaining genomic integrity. Central to this pathway is the P53 protein that is regarded as a tumor suppressor and originates from the TP53 gene. Several key discoveries regarding P53 are depicted in Figure 2, as well as its relevance in GCTs [22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][46][47][48][49][50][51][52][53][54][55][56][57][58][59]. In response to cellular stress, the P53 protein is activated and accumulates in the cell after which it mainly functions as a transcription factor that transactivates a plethora of downstream targets [49]. These target genes are key players in one of many downstream pathways including cell cycle arrest, DNA repair, apoptosis, senescence, and autophagy [51][60][61]. The P53 protein counts 393 amino acids (AA) and consists of multiple domains, starting with a transactivation domain (TAD) at the amino-terminus (N-terminus), a directly neighboring proline-rich domain (PRD), a large DNA-binding domain (DBD), a tetramerization domain (TD), and a carboxy-terminus (C-terminus) regulatory domain (REG) [62][63] (depicted in Figure 3). Once activated and accumulated in the cell, the P53 protein functions and binds DNA as a tetramer, which is facilitated by the TD [64]. When tetramerized, the DBD contains three loops, L1, L2, and L3 respectively [65]. Both L2 and L3 are bound by a zinc ion, effectively linking both loops and enabling L3 to bind to the minor groove of the DNA, while a helix in the DBD is complementary to the major groove [65][66].
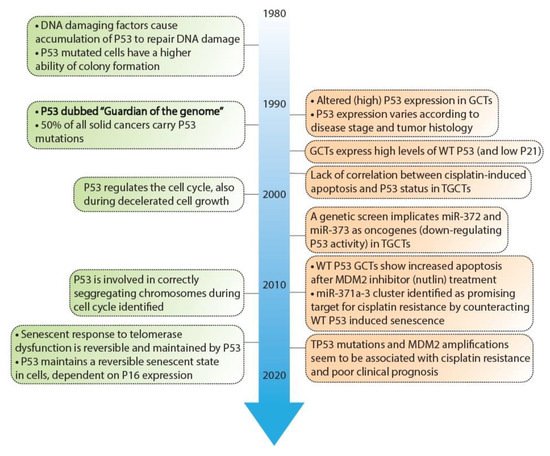
Figure 2. Timeline depicting key discoveries in P53 signaling and germ cell tumors. A timeline of the discoveries regarding P53 as a guardian of the genome (green) and studies regarding TP53 in GCT development and resistance (orange). References key discoveries P53 (green): 49–58, references key discoveries TP53 in GCT development (orange): 25–41, 59–62.

Figure 3. Schematic overview of the P53 protein. Indicated are (from N- to T-terminus) TAD: Transactivation domain, PRD: Proline-rich domain, DBD: DNA-binding domain, TD: Tetramerization domain and REG: Regulatory domain. Numbers indicate the amino acids included in each domain.
There are many different forms of cellular stress such as DNA damage that activate upstream regulators of p53, leading to the activation of the p53 pathway. For example, Ataxia-telangiectasia mutated (ATM) kinase serves as an important DNA damage sensor, as it recognizes and binds to double-strand DNA breaks and subsequently activates the P53 pathway, ultimately resulting in the activation of NHEJ or HR DNA repair mechanisms [55][67]. ATM phosphorylates histone H2AX, which is a variant of histone H2A that recruits molecules responsible for H3K9 methylation [55]. In turn, methylated H3K9 leads to the acetylation and activation of ATM [55]. Among other substrates, ATM phosphorylates and activates Checkpoint Kinase 1 and 2 (CHK1 and CHK2, respectively) [55]. In turn, CHK1 and 2 function as transcriptional activators by phosphorylating and activating the P53 protein [68]. The subsequent section will further outline several downstream targets and their related pathways including cell cycle arrest, senescence, apoptosis, and autophagy (also summarized in Figure 4).
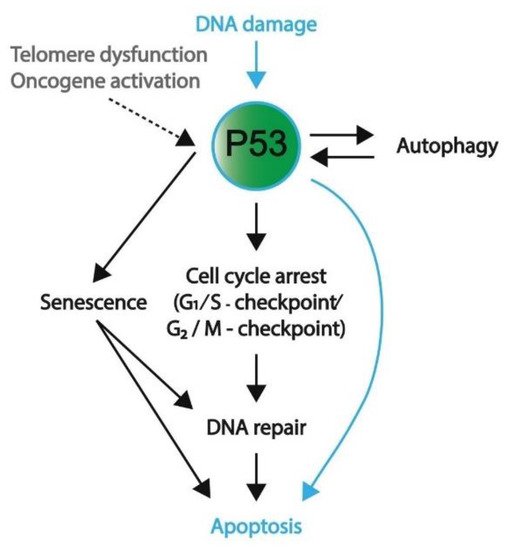
Figure 4. A schematic overview representing the favored mechanism of P53 activation in embryonic cells and somatic cells. DNA damage, both in embryonic (blue) and somatic (black) cells, will induce P53 activation. To protect genomic integrity, embryonic cells favor apoptosis over DNA repair. Conversely, somatic cells preferentially engage in cell cycle arrest and DNA repair; however, if DNA damage persists, these cells will enter apoptosis or become senescent. Furthermore, telomere dysfunction and oncogene activation are other means to activate P53 signaling (mostly in somatic cells). In addition to apoptosis, P53 activation and its downstream pathways can result in autophagy, senescence, and cell cycle arrest.
3.1. Induction of Cell Cycle Arrest
As indicated, DNA damage leads to the accumulation of P53, which induces cell cycle arrest, and subsequent DNA repair cell cycle arrest can occur at both the G1/S and G2/M checkpoints and is mostly effectuated by P21 (CDKN1A), which is a target of P53 [69]. Activated P53 binds to the promotor of P21 and initiates its transcription [70][71]. P21 inhibits the function of cyclin-dependent kinases (CDKs) CDK2 and CDK4/6 present at the G1/S transition, which results in the hypomethylation of pRB-related proteins p107 and p130 [69][72]. The transcription of cell cycle-promoting genes will subsequently be repressed, inducing a temporary block of cell cycle arrest that enables the cell to initiate DNA repair mechanisms.
3.2. Senescence or Apoptosis as a Final Measure to Protect Genomic Integrity
Chronic cellular stress signals such as telomere dysfunction, persisting DNA damage, and oncogene activation result in prolonged P53 activation and subsequent P21 expression, which in turn may induce cellular senescence or apoptosis (the latter of which is the main outcome in ES cells and GCTs) [72]. In contrast to cell cycle arrest, senescence is a stable state of arrest, and both processes are initiated by P53, again illustrating the tumor-suppressive functions of this protein [55][72]. Conversely, a senescent cell is able to re-enter the cell cycle after loss of P53 and has been shown to contribute to tumorigenesis, as it results in fast cell proliferation [27][28][73]. Prolonged P21 expression eventually induces the expression of P16INK16A (hereafter referred to as P16), which similarly functions as a CDK inhibitor for CDK4/6, thereby indirectly inhibiting pRb-family members [72]. P16 expression is known to be regulated by epigenetic factors including DNMT3, which facilitates de novo methylation of the P16 promotor, inhibiting its expression, which is maintained by DNMT1 [55]. Demethylation of the promotor consequently induces P16 expression [55]. In addition to P16, senescence is also regulated by P53-independent pathways including NF-κb, which is responsible for the expression of proinflammatory cytokines that are secreted during senescence/this cellular state [55][72]. Notably, senescent cells are non-responsive to apoptotic signals, which is demonstrated by the continued activation of cAMP response element-binding (CREB), which functions as a transcription factor for the anti-apoptotic BCL-2 protein [55]. Alternatively, persisting cellular stress signals may induce apoptotic pathways, and further information of these pathways is described in Box 1.
3.3. Autophagy
Lastly, P53 can induce autophagy, which underlines its additional role in cellular metabolism. Autophagy enables cells to maintain homeostasis by the removal and recycling of (faulty) organelles [74]. Recycling results in an increase of anabolic intermediates that can subsequently be used in multiple pathways [74][75]. There are many connections between P53 and autophagy, as illustrated in Figure 5, as autophagy is able to inactivate P53 through different mechanisms such as proteosomal degradation [76]. In addition, the autophagy protein ATG7 has been reported to bind P53 and activate the transcription of P21, resulting in cell cycle arrest [74]. Conversely, P53 promotes the transcription of genes involved in autophagy, which suggests a negative feedback loop between the autophagy proteins and P53 [74]. Additionally, a more indirect crosstalk between p53 and autophagy via the mTOR pathway is known to occur in multiple tissue types including skeletal muscle, heart, white fat, liver, and kidney tissue [33]. Here, P53 promotes the transcription of negative regulators of the IGF–AKT–mTOR pathway, resulting in higher levels of BECLIN-1, which is part of the phosphatidyl inositol-3 kinase (PI3K) complex and plays an important role in the formation of autophagosomes [77][78]. Notably, a loss of BECLIN-1 results in an increased risk of tumorigenesis, which underlines its importance in autophagy and maintaining genomic integrity [78].
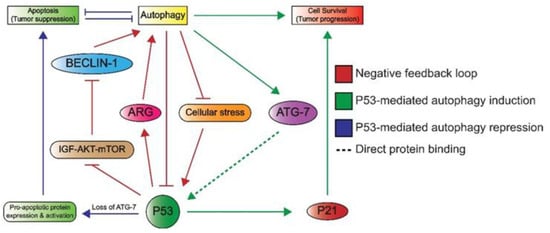
Figure 5. An overview of the crosstalk between P53 and autophagy. Autophagy can repress P53 through the inhibition of upstream activating proteins as well as P53 itself. This forms a negative feedback loop (red) as P53 induces autophagy through the inhibition of the IGF–AKT–mTOR pathway and subsequent increase in BECLIN-1 levels, which plays a role in autophagosome formation. P53 also induces the expression of autophagy-related genes (ARG). Autophagy induces cell survival, which is effectuated by the binding of ATG7 to P53, ultimately leading to P53-dependent P21 expression and subsequent cell cycle arrest (green). The loss of ATG-7 results in apoptosis (blue), as P53 induces the expression and activation of pro-apoptotic proteins. Apoptosis and autophagy have mutually inhibitory effects on one another.
References
- Juan, H.-C.; Lin, Y.; Chen, H.-R.; Fann, M.-J. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 2016, 23, 1038–1048.
- Kobayashi, T.; Surani, M.A. On the origin of the human germline. Development 2018, 145.
- Oosterhuis, J.W.; Looijenga, L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 2019, 19, 522–537.
- Dvash, T.; Ben-Yosef, D.; Eiges, R. Human Embryonic Stem Cells as a Powerful Tool for Studying Human Embryogenesis. Pediatr. Res. 2006, 60.
- Vazin, T.; Freed, W.J. Human embryonic stem cells: Derivation, culture, and differentiation: A review. Restor. Neurol. Neurosci. 2010, 28, 589–603.
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179.
- Liu, L.; Bailey, S.M.; Okuka, M.; Muñoz, P.; Li, C.; Zhou, L.; Wu, C.; Czerwiec, E.; Sandler, L.; Seyfang, A.; et al. Telomere lengthening early in development. Nat. Cell Biol. 2007, 9.
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019, 21, 1060–1067.
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline genome protection: Implications for gamete quality and germ cell tumorigenesis. Andrology 2019, 7, 516–526.
- Filion, T.M.; Qiao, M.; Ghule, P.N.; Mandeville, M.; Van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Altieri, D.C.; Stein, G.S. Survival Responses of Human Embryonic Stem Cells to DNA Damage. J. Cell. Physiol. 2009, 220, 586–592.
- Magnúsdóttir, E.; Gillich, A.; Grabole, N.; Surani, M.A. Combinatorial control of cell fate and reprogramming in the mammalian germline. Curr. Opin. Genet. Dev. 2012, 22, 466–474.
- Greenberg, M.V.C.; Bourchis, D. The diverse roles of DNA methylation in mammalian development and disease. Nature 2019, 20.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872.
- González, F.; Boué, S.; Carlos Izpisúa Belmonte, J. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011.
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors HHS Public Access. Cancer Drug Resist. 2019, 2, 580–594.
- Van Echten, J.; Sleijfer, D.T.; Wiersema, J.; Schraffordt Koops, H.; Oosterhuis, J.W.; De Jong, B. Cytogenetics of Primary Testicular Nonseminoma, Residual Mature Teratoma, and Growing Teratoma Lesion in Individual Patients. Cancer Genet. Cytogenet. 1997, 96, 1–6.
- Van Echten, J.; Van Der Vloedt, W.S.; Sleijfer, D.T.; Oosterhuis, J.W.; De Jong, B. Comparison of the Chromosomal Pattern of Primary Testicular Nonseminomas and Residual Mature Teratomas after Chemotherapy. Cancer Genet. Cytogenet. 1997, 99, 59–67.
- Jacobsen, C.; Honecker, F. Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology 2015, 3, 111–121.
- Bártková, J.; Meyts, E.R.-D.; Skakkebaek, N.E.; Lukas, J.; Bartek, J.; Meyts, R.-D.E. Deregulation of the G1/S-phase control in human testicular germ cell tumours. APMIS 2003, 111, 252–266.
- Bártková, J.; Thullberg, M.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Bartek, J. Cell cycle regulators in resticular cancer: Loss of P18INK4c marks progression from carcinoma in situ to invasive germ cell tumours. Int. J. Cancer 2000, 85, 370–375.
- Romano, F.J.; Rossetti, S.; Conteduca, V.; Schepisi, G.; Cavaliere, C.; Di Franco, R.; La Mantia, E.; Castaldo, L.; Nocerino, F.; Ametrano, G.; et al. Role of DNA repair machinery and p53 in the testicular germ cell cancer: A review. Oncotarget 2016, 7, 85641–85649.
- Spierings, D.C.; Ge De Vries, E.; Stel, A.J.; Te Rietstap, N.; Vellenga, E.; De Jong, S. Low p21 Waf1/Cip1 protein level sensitizes testicular germ cell tumor cells to Fas-mediated apoptosis. Oncogene 2004, 23, 4862–4872.
- Hong, Y.; Cervantes, R.B.; Tichy, E.; Tischfield, J.A.; Stambrook, P.J. Protecting genomic integrity in somatic cells and embryonic stem cells. Mutat. Res. 2007, 614, 48–55.
- Datta, M.W.; Macri, E.; Signoretti, S.; Renshaw, A.A.; Loda, M. Transition from In Situ to Invasive Testicular Germ Cell Neoplasia is Associated with the Loss of p21 and Gain of mdm-2 Expression. Mod. Pathol. 2001, 14, 437–442.
- Kerley-Hamilton, J.S.; Pike, A.M.; Li, N.; Direnzo, J.; Spinella, M.J. A p53-dominant transcriptional response to cisplatin in testicular germ cell tumor-derived human embyronal carcinoma. Oncogene 2005, 24, 6090–6100.
- Spierings, D.; De Vries, E.; Vellenga, E.; De Jong, S. Loss of drug-induced activation of the CD95 apoptotic pathway in a cisplatin-resistant testicular germ cell tumor cell line. Cell Death Differ. 2003, 10, 808–822.
- Spierings, D.C.; Ge De Vries, E.; Vellenga, E.; De Jong, S. The attractive Achilles heel of germ cell tumours: An inherent sensitivity to apoptosis-inducing stimuli. J. Pathol. 2003, 200, 137–148.
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. P53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to Cisplatin. PLoS ONE 2011, 6, e19198.
- Bártková, J.; Bártek, J.; Lukáŝ, J.; Vojtêŝek, B.; Staŝková, Z.; Rejthar, A.; Kovarík, J.; Midgley, C.A.; Lane, D.P. p53 Protein alterations in human testicular cancer including pre-invasive intratubular germ-cell neoplasia. Int. J. Cancer 1991, 49, 196–202.
- Burger, H.; Nooter, K.; Boersma, A.W.M.; Kortland, C.J.; Stoter, G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int. J. Cancer 1997, 73, 592–599.
- Burger, H.; Nooter, K.; Boersma, A.W.M.; van Wingerden, K.E.; Looijenga, L.H.J.; Jochemsen, A.G.; Stoter, G. Distinct p53-independent apoptotic cell death signalling pathways in testicular germ cell tumour cell lines. Int. J. Cancer 1999, 628, 620–628.
- Kersemaekers, A.M.F.; Mayer, F.; Molier, M.; Van Weeren, P.C.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H.J. Role of P53 and MDM2 in treatment response of human germ cell tumors. J. Clin. Oncol. 2002, 20, 1551–1561.
- Ulbright, T.M.; Orazi, A.; de Riese, W.; de Riese, C.; Messemer, J.E.; Foster, R.S.; Donohue, J.P.; Eble, J.N. The correlation of P53 protein expression with proliferative activity and occult metastases in clinical stage I non-seminomatous germ cell tumors of the testis. Mod. Pathol. 1994, 7, 64–68.
- Riou, G.; Barrois, M.; Prost, S.; Terrier, M.J.; Theodore, C.; Levine, A.J. The p53 and mdm-2 genes in human testicular germ-cell tumors. Mol. Carcinog. 1995, 12, 124–131.
- Schenkman, N.S.; Sesterhenn, I.A.; Washington, L.; Weghorst, C.M.; Buzard, G.S.; Srnastava, S.; Moul, J.W.; PRIIDyn, B. Increased p53 protein does not correlate to p53 gene mutations in microdissected human testicular germ cell tumors. J. Urol. 1995, 154, 617–621.
- Guillou, L.; Estreicher, A.; Chaubert, P.; Hurlimann, J.; Kurt, A.-M.; Metthez, G.; Iggo, R.; Gray, A.C.; Jichlinski, P.; Leisinger, H.-J.; et al. Germ cell tumors of the testis overexpress Wild-Type P53. Am. J. Pathol. 1996, 149, 1221–1228.
- Lutzker, S.G. P53 tumour suppressor gene and germ cell neoplasia. APMIS 1998, 106, 85–89.
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. 2016, 34, 4000–4007.
- Koster, R.; Di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; Van Den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; De Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120.
- Koster, R.; van Vugt, M.A.; Timmer-Bosscha, H.; Gietema, J.A.; de Jong, S. Unravelling mechanisms of cisplatin sensitivity and resistance in testicular cancer. Expert Rev. Mol. Med. 2013, 15, 2013.
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.A.; De Jong, S. Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011, 2.
- Lobo, J.; Alzamora, M.A.; Guimarães, R.; Cantante, M.; Lopes, P.; Braga, I.; Maurício, J.; Jerónimo, C.; Henrique, R. p53 and MDM2 expression in primary and metastatic testicular germ cell tumors: Association with clinical outcome. Andrology 2020, 8, 1233–1242.
- Ménézo, Y.; Dale, B.; Cohen, M. DNA damage and repair in human oocytes and embryos: A review. Zygote 2010, 18, 357–365.
- Xu, Y. A New Role for p53 in Maintaining Genetic Stability in Embryonic Stem Cells. Cell Cycle 2005, 363, 363–364.
- Lin, T.; Chao, C.; Saito, I.; JMazur, S.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165.
- Maltzman, W.; Czyzyk, L. UV Irradiation Stimulates Levels of p53 Cellular Tumor Antigen in Nontransformed Mouse Cells. Mol. Cell. Biol. 1984, 4, 1689–1694.
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 Protein in the Cellular Response to DNA Damage1. Cancer Res. 1991, 51, 6304–6311.
- Baker, S.J.; Markowitz, S.; Fearon, E.R.; Willson, J.K.V.; Vogelstein, B. Suppression of Human Colorectal Carcinoma Cell Growth by Wild-Type p53. Science 1990, 249, 912–915.
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 661–663.
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 Tumor Suppressor Gene: Clues to Cancer Etiology and Molecular Pathogenesis. Cancer Res. 1994, 54, 4855–4878.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Schvartzman, J.M.; Duijf, P.H.G.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714.
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170.
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222.
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28.
- Voorhoeve, P.M.; Le Sage, C.; Schrier, M.; Gillis, A.J.M.; Stoop, H.; Nagel, R.; Liu, Y.-P.; Van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates miRNA-372 and miRNA-373 As Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181.
- Dieckmann, K.-P.; Spiekermann, M.; Balks, T.; Flor, I.; Lö Ning, T.; Bullerdiek, J.; Belge, G. MicroRNAs miR-371-3 in serum as diagnostic tools in the management of testicular germ cell tumours. Br. J. Cancer 2012, 107.
- Dieckmann, K.P.; Radtke, A.; Spiekermann, M.; Balks, T.; Matthies, C.; Becker, P.; Ruf, C.; Oing, C.; Oechsle, K.; Bokemeyer, C.; et al. Serum Levels of MicroRNA miR-371a-3p: A Sensitive and Specific New Biomarker for Germ Cell Tumours. Eur. Urol. 2017, 71, 213–220.
- Dieckmann, K.-P.; Radtke, A.; Geczi, L.; Matthies, C.; Anheuser, P.; Eckardt, U.; Sommer, J.; Zengerling, F.; Trenti, E.; Pichler, R.; et al. Serum Levels of MicroRNA-371a-3p (M371 Test) as a New Biomarker of Testicular Germ Cell Tumors: Results of a Prospective Multicentric Study. J. Clin. Oncol. 2019, 37, 1412–1423.
- Helton, E.S.; Chen, X. p53 Modulation of the DNA Damage Response. J. Cell. Biochem. 2007, 100, 883–896.
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908.
- Marine, J.-C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006, 13, 927–934.
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897.
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6.
- Butler, J.S.; Loh, S.N. Structure, Function, and Aggregation of the Zinc-Free Form of the p53 DNA Binding Domain. Biochemistry 2003, 42, 2396–2403.
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a p53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic. Science 1994, 265, 346–355.
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanoví, M. The ATM signaling network in development and disease. Front. Genet. 2013, 4.
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20.
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132.
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Edward Mercer, W.; Kinzler, K.W.; Vogelstein, B. WAF1, a Potential Mediator of p53 Tumor Suppression. Cell 1993, 75, 817–825.
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-Interacting Protein Cip1 Is a Potent Inhibitor of G1 Cyclin-Dependent Kinases. Cell 1993, 75, 805–816.
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6.
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730.
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393.
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348.
- Choudhury, S.; Kolukula, V.K.; Preet, A.; Albanese, C.; Avantaggiati, M.L. Dissecting the pathways that destabilize mutant p53: The proteasome or autophagy? Cell Cycle 2013, 12, 1022–1029.
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The Regulation of AMPK B1, TSC2, and PTEN Expression by p53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-mTOR Pathways. Cancer Res. 2007, 67, 3043–3053.
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
03 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No