 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Maria Reuss | + 6052 word(s) | 6052 | 2021-06-01 04:09:37 | | | |
| 2 | Vivi Li | Meta information modification | 6052 | 2021-06-03 08:40:41 | | |
Video Upload Options
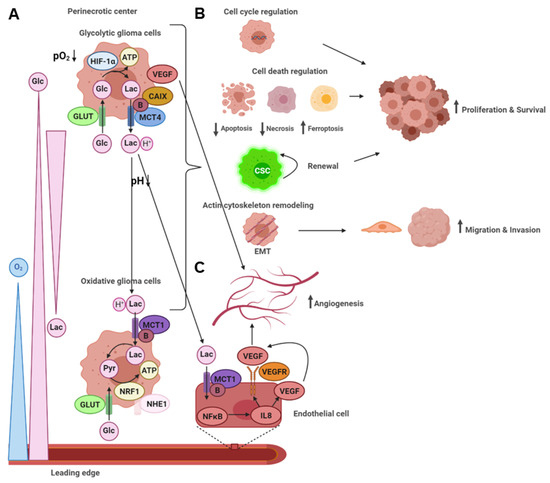
Malignant glioma represents a fatal disease with a poor prognosis and development of resistance mechanisms against conventional therapeutic approaches. The distinct tumor zones of this heterogeneous neoplasm develop their own microenvironment, in which subpopulations of cancer cells communicate. Adaptation to hypoxia in the center of the expanding tumor mass leads to the glycolytic and angiogenic switch, accompanied by upregulation of different glycolytic enzymes, transporters, and other metabolites. These processes render the tumor microenvironment more acidic, remodel the extracellular matrix, and create energy gradients for the metabolic communication between different cancer cells in distinct tumor zones. Escape mechanisms from hypoxia-induced cell death and energy deprivation are the result. The functional consequences are more aggressive and malignant behavior with enhanced proliferation and survival, migration and invasiveness, and the induction of angiogenesis.
1. Introduction
2. Lactic Acid Metabolism within the Tumor Microenvironment
2.1. Lactic Acid Production—A Hallmark of Glycolytic Cancer Cells
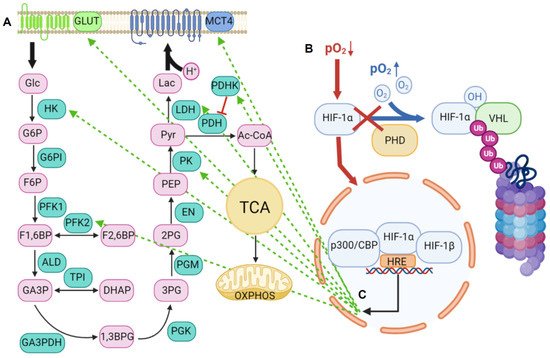
2.2. The Role of MCTs in Lactic Acid Metabolism
2.2.1. Structure and Function of MCTs
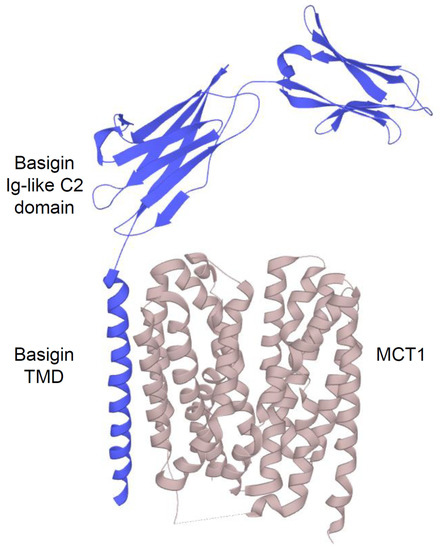
2.2.2. An acidic Tumor Microenvironment and the Metabolic Symbiosis Model
3. The Role of miRNAs in the Glycolytic Switch
| miRNA | Expression in Glioma | Targeted by | Targets | Effects in Glioma | Literature |
|---|---|---|---|---|---|
| miR-1 | Downregulated | - | Annexin A2 | Decreases proliferation, invasion, and angiogenesis in glioma cells and xenografts | [65] |
| miR-9 | Overexpressed | CREB | CREB, NF1 |
Decreases proliferation and increases migration in glioma cells | [66] |
| miR-29a | Downregulated | - | PDGFC, PDGFA | Decreases proliferation, cell viability, migration, and invasion in glioma cells, and tumor growth in xenografts | [67] |
| miR-95-3p | Downregulated | - | CELF2 | Decreases proliferation, cell viability, and invasion in glioma cells | [68] |
| miR-124 | Downregulated | - | SNAl2 | Decreases proliferation and invasion in glioma cells and tumor growth in xenografts | [69] |
| miR-134 | Downregulated | - | KRAS, STAT5B |
Decreases proliferation and cell viability in glioma and glioma stem cells and tumor growth in xenografts | [70] |
| miR-145 | Downregulated | - | - | Decreases migration and invasion in glioma cells | [71] |
| miR-148a | Overexpressed | - | MIG6, BIM |
Increases proliferation, cell viability, migration, and invasion in glioma cells, and tumor growth in xenografts | [72] |
| miR-150 | Overexpressed | - | VHL | Increases glucose uptake, lactate secretion, and proliferation in glioma cells, and tumor growth in xenografts | [64] |
| miR-181b | Downregulated | - | SP1 | Decreases glucose uptake and proliferation in glioma cells and tumor growth in xenografts | [73] |
| miR-181d | Downregulated | - | KRAS, Bcl-2 |
Decreases proliferation and cell viability in glioma cells and tumor growth in xenografts | [74] |
| miR-203 | Downregulated | - | - | - | [75] |
| miR-338-3p | Downregulated | circSMO742 &SMO | - | Decreases proliferation, cell viability, migration, and invasion in glioma cells | [76] |
| miR-351 | Overexpressed | - | NAIF1 | Increases cell viability, migration, and invasion in glioma cells | [77] |
| miR-378e | Downregulated | circNFIX | RPN2 | Decreases glucose uptake, lactate secretion, cell viability, migration, and invasion in glioma cells | [78] |
| miR-423-5p | Overexpressed | - | ING-4 | Increases proliferation, invasion, angiogenesis, and temozolomide resistance in glioma cells and tumor growth and invasion in xenografts | [79] |
| miR-432-5p | Downregulated | - | RAB10 | Decreases glucose uptake, lactate secretion, invasion, and proliferation in glioma cells | [80] |
| miR-451 | Downregulated | - | - | Increases cell viability and decreases invasion in glioma cells | [62] |
| miR-451 | Downregulated | - | CAB39 | Decreases proliferation, invasion, and migration in glioma cells and tumor growth in xenografts | [63] |
| miR-451 | Downregulated in low glucose level glioma cells | - | - | Decreases migration in glioma cells andinvasion in xenografts, increases sensitivity to temozolomide treatment in glioma cells | [60] |
| miR-451 | Downregulated | - | - | Decreases proliferation, cell viability, and invasion in glioma cells | [81] |
| miR-451 | Downregulated | lncRNA LSINCT5 | CAB39 | Decreases glycolysis, cell viability, invasion, and migration in glioma cells | [61][82] |
| miR-495 | Downregulated | - | GLUT1 | Decreases glucose uptake and lactate secretion in glioma cells | [58] |
| miR-663 | Downregulated | - | PIK3CD | Decreases proliferation and invasion in glioma cells | [83] |
4. The Acidic Tumor Microenvironment in IDH wt versus IDH Mutant Gliomas
In recent years, the major prognostic relevance of IDH mutation status has been recognized, heralding the era of the integrated histological-molecular approach for diagnosing brain tumors [7][8]. Missense mutations in the IDH1 or IDH2 gene lead to the replacement of positively charged arginine residues by non-charged, polar amino acids, such as histidine or cysteine [84][85]. The resultant impairment of hydrogen bond formation with the carboxy sites reduces the affinity for isocitrate. It increases the preference for nicotinamide adenine dinucleotide phosphate (NADPH) [86]. Since most cancer cells harbor heterozygous IDH mutations, IDH heterodimers reveal, on one hand, the wt form, converting isocitrate and NADP+ into α-ketoglutarate (KG), carbon dioxide (CO2), and NADPH. On the other hand, the mutant form displays neomorphic activity, converting α-KG into the enantiomer D-2-hydroxyglutarate (D-2-HG) in an NADPH-dependent manner (Figure 4).
D-2-HG inhibits α-KG dependent enzymes and leads to a hypermethylation phenotype [87][88][89][90]. Since IDH is a crucial enzyme in the TCA cycle, other metabolic pathways in the tumor microenvironment may be impacted by epigenetic regulation.
Initially, IDH1 mutated glioma cells have been reported to produce lower amounts of α-KG in favor of D-2-HG, leading to HIF-1α overexpression. Thus, IDH1 has been suggested to act as a tumor suppressor, which, when inactivated by mutation, contributes to tumorigenesis in part by induction of the HIF-1α pathway [91].
In contrast, another study has shown that D-2-HG stimulates PHDs, leading to reduced HIF-1α levels in IDH mutant cells [92]. Simultaneous pH and oxygen-sensitive MRI have revealed higher acidity and higher hypoxia in IDH wt than IDH mutant gliomas, correlating to higher HIF-1α expression and increased proliferation index [93]. Accordingly, HIF-1α target genes, including many glycolytic genes, are downregulated in IDH mutant compared to IDH wt gliomas [118]. Further gene expression analysis has revealed a glycolytic phenotype for IDH wt gliomas. In contrast, IDH mutant gliomas overexpress genes encoding for TCA cycle involved enzymes [94]. This finding has been confirmed by in vitro experiments, showing that IDH mutant HCT 116 cells have higher basal respiration rates than IDH wt cells. Likewise, lactate levels are significantly increased in IDH wt gliomas. In contrast, IDH mutant cells exhibit reduced hyperpolarized lactate production along with decrease of LDH as well as MCT1 and MCT4 levels [95][96][97][98].
Therefore, epigenetic silencing of glycolytic switch-related genes may explain why IDH mutant gliomas exhibit slower proliferation and less aggressive behavior than IDH wt gliomas [97].
On the other hand, acquisition of the Warburg phenotype has been associated with the CpG island methylator phenotype (G-CIMP) in IDH mutant astrocytoma, displaying more aggressive behavior than IDH mutant oligodendroglioma [99]. Consistently, D-2-HG overproduction inhibits OXPHOS in IDH1R132H mutant glioma cells, thereby reducing ATP levels and activating the AMPK pathway [100]. AMPK-mediated inhibition of the mammalian target of rapamycin (mTOR) signaling decreases protein synthesis, making IDH mutant glioma cells vulnerable to synthetic lethality through inhibition of B cell lymphoma-extra large (Bcl-xL). This finding is consistent with a prior study, showing that D-2-HG inhibits ATP synthase and that IDH1R132H mutant cells display reduced ATP levels and mTOR signaling [101].
However, another study has suggested that D-2-HG leads to mTOR activation in IDH1R132H mutant cells by disinhibition via the lysine demethylase (KDM)4A [102].
The PIK3/AKT/mTOR pathway is known to enhance glucose uptake via GLUT1 and to control the glycolytic flux through regulation of glycolytic enzymes without affecting the rate of OXPHOS [103][104][105]. Under hypoxic conditions, mTOR has been suggested to activate HIF-1α, thereby promoting the glycolytic switch [106].
Regarding these intricate signaling pathways, it is conceivable that IDH mutation can regulate glycolysis connected to OXPHOS in glioma cells. When undergoing the Warburg effect or under hypoxic conditions, IDH mutant cells may also promote the glycolytic switch. Therefore, the effects are likely highly dependent on the cell status and predominant microenvironment in IDH mutant gliomas, which may vary considerably between different grades. However, the relation between IDH mutation, D-2-HG, and HIF-1α is still controversial and needs further clarification.
It is clear that IDH mutations, redirecting carbon metabolites away from the TCA cycle towards D-2-HG production, decrease oxidative metabolism and shift the redox potential to a more oxidated state. The resultant increase in oxidative stress has been related to the enhanced sensitivity of IDH mutant cells to chemotherapy [107][108][109][110][111].
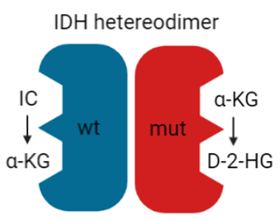
Figure 4. IDH heterodimer. IDH wild-type (wt; blue) and mutant (mut; red) monomers form a catalytically active heterodimer. IDH wt converts isocitrate (IC) to α-KG while IDH mut converts α-KG to D-2-HG.
5. Implications for Diagnostics and Therapy
Malignant glioma is a fatal neoplasm with a very poor prognosis despite advances in surgical techniques and combined treatment with radio-chemotherapy [4][5][6]. In this review, it has been highlighted that gliomas with glycolytic and angiogenic switch tumor microenvironments display more aggressive and malignant behavior. Therefore, identification of these specific tumor regions in vivo and local, specific targeting of glycolytic glioma cells may give rise to new therapeutic approaches.
5.1. Diagnostic Approaches for Identifying Glycolytic Tumor Regions
Hypoxic brain regions with low pH and elevated lactate levels can be identified in patients using MRI and positron emission tomography (PET) techniques [51][53][112][113]. Radiotracers for MCT and CAIX imaging in brain gliomas are also under development [114][115]. Furthermore, MRSI has been indicated for the monitoring of metabolic responses to treatments via the lactate-to-pyruvate-ratio, which is more sensitive than evaluation of tumor growth by conventional MRI [97]. Interestingly, MRSI monitoring of the lactate-to-pyruvate-ratio during treatment of glioma cells with the histone deacetylase (HDAC) inhibitor vorinostat has revealed a significant decrease, accompanied by upregulation of MCT4 and MCT1 [116]. Therefore, MRSI may serve as an important monitoring tool to detect resistance mechanisms during the therapy of malignant glioma.
5.2. Glycolytic Players as Targets in Glioma Therapy
As mentioned above, inhibitor treatment of MCTs in glioma cells significantly reduces proliferation and survival, migration and invasiveness, and the induction of angiogenesis [43][53][55][117][118][119]. General MCT inhibition may be problematic due to physiological expression in skeletal muscle and brain astrocytes, where astrocyte-neuron lactate shuttling is crucial for memory formation [30][120][121]. However, programmed orthotopic administration of the MCT inhibitor CHC by osmotic pumps into gliomas implanted into rats has been shown to substantially decrease invasion and to lead to necrosis within the tumor bed [122]. Importantly, no neurological side effects have been observed. Likewise, the small molecule acriflavine, targeting the binding between MCT4 and its chaperone basigin, has been revealed to inhibit the growth and self-renewal potential of glioma neurospheres, especially under hypoxia [123]. In stem cell-derived xenograft mice, acriflavine markedly reduces tumor progression and vascularization by VEGF inhibition. Vice versa, hypoxic and glycolytic markers like HIF-1α, CAIX, GLUT1, and MCT1 are upregulated in glioma cells under bevacizumab treatment [124]. This may explain the resistance that gliomas acquire during therapy. In contrast, inhibition of CAIX or glucose uptake enhances the effects of bevacizumab [125][124]. Likewise, pretreatment of glioma cells targeting CAIX or MCT4 increases sensitivity to subsequent radio-chemotherapy [126][127]. Moreover, a combination of the conventional chemotherapeutic agent temozolomide with the CAIX inhibitor acetazolamide significantly enhances cell death of glioma cells and glioma stem cells under hypoxic conditions [128].
Furthermore, HIF-1α has been suggested as a potential therapeutic target using small interference (si)RNA, packaged in a novel surfactant-based nucleic acid carrier and delivered into gliomas of an in vivo orthotopic mouse model by osmotic pumps [129]. HIF-1α silencing is associated with the downregulation of the transcriptional targets GLUT1, CAIX, and VEGF. It has reduced the tumor volume by 79% after 50 days of daily treatment. Proliferation index and microvascular density have also been significantly lower.
Additionally, other glycolytic inhibitors like 3-bromopyruvate (3-BP), which is structurally related to lactate and pyruvate, the PFK inhibitor citrate and enolase inhibitor sodium fluoride, have been proposed as energy depletion therapy in glioma cells [130]. 3-BP induces caspase-dependent cell death and blocks migration of glioma cells promoted by lactate. Notably, 3-BP and citrate show synergistic effects in decreasing glioma cell viability.
Besides apoptosis and necrosis, ferroptosis is a recently proposed iron-dependent mechanism of cell death [131]. Interestingly, MCT4 overexpressing glioma cells show sensitivity to ferroptosis compared to normal or reduced MCT4 expression levels [43]. This finding indicates another selective therapeutic approach for locally targeting exclusively MCT4 overexpressing cells in malignant glioma, which warrants further exploration.
Finally, metabolic changes in the hypoxia-induced tumor microenvironment under EGFR and mTOR inhibition, leading to adverse effects, have suggested the exact opposite approach [132]. Instead, mTOR activation by suppressing its physiological inhibitor tuberose sclerosis complex (TSC)2 causes hypoxia-induced glioma cell death by earlier ATP depletion and reactive oxygen species (ROS) production. Identification of the underlying mechanisms has revealed enhanced oxygen consumption due to the upregulation of genes involved in OXPHOS and increased metabolites of the pentose phosphate pathway due to upregulation of the rate-limiting enzyme G6P. This finding may also be beneficial in combination with hypoxia-inducing therapies like bevacizumab treatment.
In summary, exploiting the glycolytic switch for selective tumor therapy offers a broad range of novel possibilities, which malignant glioma patients may profit from soon.
5.3. Impact of the Glycolytic Phenotype on Tumor Immunity and Immunotherapy
In glioma, expression of immune checkpoint genes is associated with more aggressive tumor behavior and worse prognosis [133]. Interestingly, genomic analysis has revealed an epigenetic link between glycolytic and immune checkpoint gene expression in low-grade glioma [134]. In this study, the IDH wt cluster displayed lower levels of LDHA promoter methylation and a higher LDHA/LDHB expression ratio. This genotype was accompanied by less promotor methylation of the immune inhibitory molecule programmed cell death ligand (PDL)1/2 and thus higher PDL1/2 expression levels. In contrast, IDHR132H induction decreased promotor histone (H)3K4 triple methylation (me3) for LDHA and PDL1/2. Crosstalk between the immune checkpoint and metabolic pathways may profoundly impact tumor cell evasion from immune system recognition. For instance, in glioma, uncoupling protein (UCP)2 has been proposed to link the glycolytic switch to dampened immune response [135].
Active immune cells also exhibit the Warburg effect to fulfill their energy demands. For example, quiescent naïve T lymphocytes use OXPHOS, whereas activation induces the glycolytic switch in these cells [136][137]. Therefore, nutrient competition may be one metabolic mechanism involved in tumor cell evasion from the immune system. Indeed, glycolytic tumors with overexpression of GLUT1 and LDHA and enhanced lactate secretion show an inverse correlation with infiltration of CD8+ cytotoxic T lymphocytes (CTLs) [138]. Furthermore, glycolytic tumor-infiltrating T cells exhibit fewer effector molecules, such as granzyme B and perforin, and thus reduced cytotoxicity. In a mouse sarcoma model, it has been demonstrated that increased glycolysis and thus glucose consumption in tumor cells metabolically restricts T lymphocytes by reducing mTOR activity, glycolytic capacity, and interferon (IFN)-γ production in these cells [139]. Abrogation of proper T cell function is sufficient to promote tumor growth. In contrast, checkpoint blockade antibodies against CTL-associated protein (CTLA)4, PD1, and PDL1 restores glucose levels in the tumor microenvironment, T cell function, and IFN-γ production. Mechanistically, PDL1 blockade has been shown to decrease glycolysis in tumor cells by inhibiting mTOR activity and reduced the expression of glycolytic enzymes. However, rapamycin-mediated inhibition of immune checkpoint molecule induced mTOR pathway in naïve CD8+ T cells promotes the production of memory T cell precursors by persistent eomesodermin expression [140]. These cells show enhanced antigen-recall responses upon adoptive transfer and higher tumor efficacy. Consistently, it has been demonstrated that enhanced glycolysis in CTLs impairs memory T cell generation by driving them towards a terminally differentiated state [141]. Notably, these cells fail during adoptive transfer. In contrast, inhibition of glycolysis in tumor-specific CTLs increases antitumor response.
Vice versa, tumor cell glycolysis has been associated with immune resistance to adoptive T cell therapy in melanoma [142]. Highly glycolytic melanoma cells display reduced levels of interferon regulatory factor (IRF)1 and C-X-C motif chemokine ligand (CXCL)10 immunostimulatory molecules.
Furthermore, hypoxia leads to HIF-1α dependent PDL1 upregulation in tumor cells, thereby increasing resistance to CTL lysis [143]. Hypoxia-mediated PDL1 overexpression in tumor cells increases apoptosis in co-cultured T cells, which is abrogated by blocking the interaction of PDL1 and the PD1 receptor on T cells. Furthermore, treatment with glyceryl trinitrate (GTN), an agonist of nitric oxide (NO) signaling known to block HIF-1α accumulation in hypoxic cells, prevents hypoxia-mediated PDL1 overexpression and reduces T cell apoptosis. This is accompanied by decreased tumor cell immune escape from CTL-mediated lysis and reduces tumor growth, suggesting novel cancer immunotherapy to block PDL1 expression specifically in hypoxic tumor cells by administering NO mimetics.
Moreover, PD1 intrinsically expressed in melanoma cell subpopulations has been shown to promote tumorigenesis via mTOR signaling, even in mice lacking adaptive immune response [144]. In contrast, PD1 inhibition by antibodies, siRNA or mutagenesis of PD1 signaling motifs substantially reduces tumor growth.
Interestingly, it has been shown that PD1 overexpression in tumor-infiltrating T lymphocytes during prolonged antigen exposure leads to DNA methylation and is responsible for complete T cell exhaustion, which is resistant to immune checkpoint blockade mediated rejuvenation [145]. Intriguingly, the authors propose approaches for DNA methylation reprogramming, which improves T cell responses and tumor control during immune checkpoint blockade. This discovery may also give rise to developing novel strategies in glioma immunotherapy.
Besides suppressing tumor immunity via immune checkpoint modulation in glycolytic tumor cells, a direct inhibitory effect of tumor cell-derived lactic acid on CTL proliferation, cytokine production, and cytotoxicity against tumor spheroids has been shown [146]. The authors propose that high lactic acid levels within the tumor microenvironment block the export of lactic acid from T cells through MCT1, thereby disturbing their metabolism and function.
These results suggest that targeting glycolytic pathways in tumor and T cells combined with immunotherapy opens new perspectives in cancer treatment. For instance, CAIX has been demonstrated as a suitable target for selective chimeric antigen receptor (CAR) T cell therapy with a cure rate of 20% and without any systemic side effects in an in vivo glioma xenograft mouse model [147].
Besides T lymphocytes, tumor-associated macrophages (TAMs) play a crucial role in glioma immunity and glioma cell evasion [148]. Interestingly, glioma cells have been shown to secrete branched-chain ketoacids via MCT1, taken up by TAMs, where they reduce phagocytosis [149]. This finding raises the possibility of a role of MCTs in tumor immune suppression. Furthermore, upregulated CAIX leads to TAM M2 polarization, indicating an immunosuppressive phenotype in glioma [150]. M2 TAMs produce IL-1β, which in turn leads to phosphorylation of the glycolytic enzyme GA3PDH in glioma cells through PIK3-mediated activation of protein kinase (PK)Cδ [151]. Blocking of the respective factors reduces the glycolytic rate and proliferation of glioma cells. In patient-derived glioma tissue, staining for IL-1β and macrophages correlates with PKCδ and GAP3DH, and this is associated with higher glioma grade and lower overall survival.
Taken together, these findings suggest interference of cytokine crosstalk between M2 TAMs and glioma cells as a further possible treatment approach.
Finally, it has been shown that sensitization of glioma cells by low-dose administration of attenuated oncolytic measles virus Edmonston strain (MV-Edm), leading to a shift to high-rate aerobic glycolysis, to the glycolytic blocker dichloroacetate, induces substantial cell death of tumor, but not of non-tumor cells in vitro as well as in an in vivo GBM xenograft model [152]. Besides blocking bioenergetic generation, dichloroacetate enhances viral replication by abrogating mitochondrial antiviral signaling protein (MAVS)-mediated innate immune response, increasing bioenergetic consumption and oncolysis.
This somewhat uncommon approach seems to be quite powerful by combining different effects and should be considered for further investigation.
In summary, exploiting the glycolytic phenotype for combined immunotherapy with distinct components in tumors and different immune cells may constitute a promising therapeutic approach. Due to the combined targeting of various effectors, is likely more powerful and less prone to resistance mechanisms than focusing on single mechanisms. Large and sophisticated studies are necessary to explore the complexity of such an approach in detail.
6. Conclusions and Future Perspectives
Glioma cells in the perinecrotic tumor center live under hypoxic conditions. These cancer cells undergo a glycolytic switch from OXPHOS to anaerobic glycolysis via regulation by HIF-1α, inducing upregulation of glycolytic enzymes, transporters, and VEGF. This, in turn, triggers the angiogenic switch, leading to neovascularization. Lactate, the end product of anaerobic glycolysis, is exported as lactic acid primarily via MCT4, thereby increasing acidity in the tumor microenvironment. The low extracellular pH elevated lactate levels, and other metabolites produced by the high glycolytic flux lead to metabolic gradients across the tumor microenvironment, remodel the extracellular matrix and activate signaling pathways in neighboring cells. Of note, lactic acid uptake by endothelial cells via MCT1 leads to the induction of angiogenesis. Neovascularization, migration and invasion, as well as proliferation and survival, are the functional consequences, rendering malignant gliomas even more aggressive and resistant to established treatment regimens.
However, the glycolytic switch can also be exploited for developing novel therapeutic approaches by taking advantage of the weak points in glycolytic cells, such as sensitivity to oxidative stress and hypoxia-induced cell death. Modern therapeutic concepts or their combination with conventional treatment regimens or immunotherapy may overcome cancer cell resistance to achieve a better prognosis for malignant glioma patients. The development of selective targeting, applicable to glioma patients without severe side effects, especially for other glycolytic cells like skeletal muscle or brain astrocytes, represents the crucial challenge for future studies.
References
- Rasmussen, B.K.; Hansen, S.; Laursen, R.J.; Kosteljanetz, M.; Schultz, H.; Norgard, B.M.; Guldberg, R.; Gradel, K.O. Epidemiology of glioma: Clinical characteristics, symptoms, and predictors of glioma patients grade I-IV in the the Danish Neuro-Oncology Registry. J. Neurooncol. 2017, 135, 571–579.
- Davis, F.G.; Smith, T.R.; Gittleman, H.R.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Glioblastoma incidence rate trends in Canada and the United States compared with England, 1995-2015. Neuro. Oncol. 2019.
- Porter, K.R.; McCarthy, B.J.; Freels, S.; Kim, Y.; Davis, F.G. Prevalence estimates for primary brain tumors in the United States by age, gender, behavior, and histology. Neuro Oncol. 2010, 12, 520–527.
- Castro, M.G.; Cowen, R.; Williamson, I.K.; David, A.; Jimenez-Dalmaroni, M.J.; Yuan, X.; Bigliari, A.; Williams, J.C.; Hu, J.; Lowenstein, P.R. Current and future strategies for the treatment of malignant brain tumors. Pharm. Ther. 2003, 98, 71–108.
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648.
- Anton, K.; Baehring, J.M.; Mayer, T. Glioblastoma multiforme: Overview of current treatment and future perspectives. Hematol. Oncol. Clin. N. Am. 2012, 26, 825–853.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820.
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856.
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942.
- Mashima, T.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Promotion of glioma cell survival by acyl-CoA synthetase 5 under extracellular acidosis conditions. Oncogene 2009, 28, 9–19.
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219.
- Wu, R.; Racker, E. Regulatory mechanisms in carbohydrate metabolism. IV. Pasteur effect and Crabtree effect in ascites tumor cells. J. Biol. Chem. 1959, 234, 1036–1041.
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharm. 2011, 2, 49.
- Semenza, G.L.; Artemov, D.; Bedi, A.; Bhujwalla, Z.; Chiles, K.; Feldser, D.; Laughner, E.; Ravi, R.; Simons, J.; Taghavi, P.; et al. ‘The metabolism of tumours’: 70 years later. Novartis Found. Symp. 2001, 240, 251–260; discussion 260–264.
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340.
- Hirsilä, M.; Koivunen, P.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275.
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468.
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973.
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454.
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ. Res. 1995, 77, 638–643.
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162.
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185.
- Curi, R.; Newsholme, P.; Newsholme, E.A. Metabolism of pyruvate by isolated rat mesenteric lymphocytes, lymphocyte mitochondria and isolated mouse macrophages. Biochem. J. 1988, 250, 383–388.
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033.
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 1972, 60, 56–63.
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell Proteom. 2017, 16, 1906–1921.
- Branco, M.; Linhares, P.; Carvalho, B.; Santos, P.; Costa, B.M.; Vaz, R. Serum lactate levels are associated with glioma malignancy grade. Clin. Neurol. Neurosurg. 2019, 186, 105546.
- Price, N.T.; Jackson, V.N.; Halestrap, A.P. Cloning and sequencing of four new mammalian monocarboxylate transporter (MCT) homologues confirms the existence of a transporter family with an ancient past. Biochem. J. 1998, 329, 321–328.
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343, 281–299.
- Saier, M.H. Computer-aided analyses of transport protein sequences: Gleaning evidence concerning function, structure, biogenesis, and evolution. Microbiol. Rev. 1994, 58, 71–93.
- Poole, R.C.; Halestrap, A.P. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am. J. Physiol. 1993, 264, C761–C782.
- Halestrap, A.P. The monocarboxylate transporter family--Structure and functional characterization. Iubmb Life 2012, 64, 1–9.
- Poole, R.C.; Halestrap, A.P. Interaction of the erythrocyte lactate transporter (monocarboxylate transporter 1) with an integral 70-kDa membrane glycoprotein of the immunoglobulin superfamily. J. Biol. Chem. 1997, 272, 14624–14628.
- Kirk, P.; Wilson, M.C.; Heddle, C.; Brown, M.H.; Barclay, A.N.; Halestrap, A.P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000, 19, 3896–3904.
- Wilson, M.C.; Meredith, D.; Fox, J.E.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: The ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221.
- Manoharan, C.; Wilson, M.C.; Sessions, R.B.; Halestrap, A.P. The role of charged residues in the transmembrane helices of monocarboxylate transporter 1 and its ancillary protein basigin in determining plasma membrane expression and catalytic activity. Mol. Membr. Biol. 2006, 23, 486–498.
- Wilson, M.C.; Meredith, D.; Bunnun, C.; Sessions, R.B.; Halestrap, A.P. Studies on the DIDS-binding site of monocarboxylate transporter 1 suggest a homology model of the open conformation and a plausible translocation cycle. J. Biol. Chem. 2009, 284, 20011–20021.
- Guenette, R.S.; Sridhar, S.; Herley, M.; Mooibroek, M.; Wong, P.; Tenniswood, M. Embigin, a developmentally expressed member of the immunoglobulin super family, is also expressed during regression of prostate and mammary gland. Dev. Genet. 1997, 21, 268–278.
- Froberg, M.K.; Gerhart, D.Z.; Enerson, B.E.; Manivel, C.; Guzman-Paz, M.; Seacotte, N.; Drewes, L.R. Expression of monocarboxylate transporter MCT1 in normal and neoplastic human CNS tissues. Neuroreport 2001, 12, 761–765.
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro Oncol. 2013, 15, 172–188.
- Reuss, A.M.; Groos, D.; Ghoochani, A.; Buchfelder, M.; Savaskan, N. MCT4 Promotes Tumor Malignancy in F98 Glioma Cells. J. Oncol. 2021, 2021, 6655529.
- Duan, K.; Liu, Z.J.; Hu, S.Q.; Huo, H.Y.; Xu, Z.R.; Ruan, J.F.; Sun, Y.; Dai, L.P.; Yan, C.B.; Xiong, W.; et al. Lactic acid induces lactate transport and glycolysis/OXPHOS interconversion in glioblastoma. Biochem. Biophys Res. Commun. 2018, 503, 888–894.
- Grillon, E.; Farion, R.; Fablet, K.; De Waard, M.; Tse, C.M.; Donowitz, M.; Remy, C.; Coles, J.A. The spatial organization of proton and lactate transport in a rat brain tumor. PLoS ONE 2011, 6, e17416.
- Cheng, C.; Edin, N.F.; Lauritzen, K.H.; Aspmodal, I.; Christoffersen, S.; Jian, L.; Rasmussen, L.J.; Pettersen, E.O.; Xiaoqun, G.; Bergersen, L.H. Alterations of monocarboxylate transporter densities during hypoxia in brain and breast tumour cells. Cell Oncol. (Dordr.) 2012, 35, 217–227.
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037.
- Kaluz, S.; Kaluzová, M.; Liao, S.Y.; Lerman, M.; Stanbridge, E.J. Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: A one transcription factor (HIF-1) show? Biochim. Biophys. Acta 2009, 1795, 162–172.
- Ames, S.; Andring, J.T.; McKenna, R.; Becker, H.M. CAIX forms a transport metabolon with monocarboxylate transporters in human breast cancer cells. Oncogene 2020, 39, 1710–1723.
- Gerweck, L.E.; Seetharaman, K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 1996, 56, 1194–1198.
- Ferrauto, G.; Di Gregorio, E.; Auboiroux, V.; Petit, M.; Berger, F.; Aime, S.; Lahrech, H. CEST-MRI for glioma pH quantification in mouse model: Validation by immunohistochemistry. Nmr. Biomed. 2018, 31, e4005.
- Li, X.; Vigneron, D.B.; Cha, S.; Graves, E.E.; Crawford, F.; Chang, S.M.; Nelson, S.J. Relationship of MR-derived lactate, mobile lipids, and relative blood volume for gliomas in vivo. Ajnr Am. J. Neuroradiol. 2005, 26, 760–769.
- Kubelt, C.; Peters, S.; Ahmeti, H.; Huhndorf, M.; Huber, L.; Cohrs, G.; Hövener, J.B.; Jansen, O.; Synowitz, M.; Held-Feindt, J. Intratumoral Distribution of Lactate and the Monocarboxylate Transporters 1 and 4 in Human Glioblastoma Multiforme and Their Relationships to Tumor Progression-Associated Markers. Int. J. Mol. Sci. 2020, 21, 6254.
- Reichert, M.; Steinbach, J.P.; Supra, P.; Weller, M. Modulation of growth and radiochemosensitivity of human malignant glioma cells by acidosis. Cancer 2002, 95, 1113–1119.
- Miranda-Gonçalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335–46353.
- Bröer, S.; Schneider, H.P.; Bröer, A.; Rahman, B.; Hamprecht, B.; Deitmer, J.W. Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem. J. 1998, 333, 167–174.
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115.
- Nie, S.; Li, K.; Huang, Y.; Hu, Q.; Gao, X.; Jie, S. miR-495 mediates metabolic shift in glioma cells via targeting Glut1. J. Craniofac. Surg. 2015, 26, e155–e158.
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; De Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632.
- Ogawa, D.; Ansari, K.; Nowicki, M.O.; Salińska, E.; Bronisz, A.; Godlewski, J. MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy. Noncoding RNA 2019, 5, 25.
- Guo, H.; Nan, Y.; Zhen, Y.; Zhang, Y.; Guo, L.; Yu, K.; Huang, Q.; Zhong, Y. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumour. Biol. 2016, 37, 13751–13761.
- Zhao, K.; Wang, L.; Li, T.; Zhu, M.; Zhang, C.; Chen, L.; Zhao, P.; Zhou, H.; Yu, S.; Yang, X. The role of miR-451 in the switching between proliferation and migration in malignant glioma cells: AMPK signaling, mTOR modulation and Rac1 activation required. Int. J. Oncol. 2017, 50, 1989–1999.
- Nan, Y.; Guo, H.; Guo, L.; Wang, L.; Ren, B.; Yu, K.; Huang, Q.; Zhong, Y. MiRNA-451 Inhibits Glioma Cell Proliferation and Invasion Through the mTOR/HIF-1α/VEGF Signaling Pathway by Targeting CAB39. Hum. Gene Clin. Dev. 2018, 29, 156–166.
- Li, S.J.; Liu, H.L.; Tang, S.L.; Li, X.J.; Wang, X.Y. MicroRNA-150 regulates glycolysis by targeting von Hippel-Lindau in glioma cells. Am. J. Transl Res. 2017, 9, 1058–1066.
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750.
- Tan, X.; Wang, S.; Yang, B.; Zhu, L.; Yin, B.; Chao, T.; Zhao, J.; Yuan, J.; Qiang, B.; Peng, X. The CREB-miR-9 negative feedback minicircuitry coordinates the migration and proliferation of glioma cells. PLoS ONE 2012, 7, e49570.
- Yang, Y.; Dodbele, S.; Park, T.; Glass, R.; Bhat, K.; Sulman, E.P.; Zhang, Y.; Abounader, R. MicroRNA-29a inhibits glioblastoma stem cells and tumor growth by regulating the PDGF pathway. J. Neurooncol. 2019, 145, 23–34.
- Fan, B.; Jiao, B.H.; Fan, F.S.; Lu, S.K.; Song, J.; Guo, C.Y.; Yang, J.K.; Yang, L. Downregulation of miR-95-3p inhibits proliferation, and invasion promoting apoptosis of glioma cells by targeting CELF2. Int. J. Oncol. 2015, 47, 1025–1033.
- Xia, H.; Cheung, W.K.C.; Ng, S.S.; Jiang, X.; Jiang, S.; Sze, J.; Leung, G.K.K.; Lu, G.; Chan, D.T.M.; Bian, X.W.; et al. Loss of brain-enriched miR-124 microRNA enhances stem-like traits and invasiveness of glioma cells. J. Biol. Chem. 2012, 287, 9962–9971.
- Zhang, Y.; Kim, J.; Mueller, A.C.; Dey, B.; Yang, Y.; Lee, D.H.; Hachmann, J.; Finderle, S.; Park, D.M.; Christensen, J.; et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. 2014, 21, 720–734.
- Lee, S.J.; Kim, S.J.; Seo, H.H.; Shin, S.P.; Kim, D.; Park, C.S.; Kim, K.T.; Kim, Y.H.; Jeong, J.S.; Kim, I.H. Over-expression of miR-145 enhances the effectiveness of HSVtk gene therapy for malignant glioma. Cancer Lett. 2012, 320, 72–80.
- Kim, J.; Zhang, Y.; Skalski, M.; Hayes, J.; Kefas, B.; Schiff, D.; Purow, B.; Parsons, S.; Lawler, S.; Abounader, R. microRNA-148a is a prognostic oncomiR that targets MIG6 and BIM to regulate EGFR and apoptosis in glioblastoma. Cancer Res. 2014, 74, 1541–1553.
- Yin, J.; Shi, Z.; Wei, W.; Lu, C.; Wei, Y.; Yan, W.; Li, R.; Zhang, J.; You, Y.; Wang, X. MiR-181b suppress glioblastoma multiforme growth through inhibition of SP1-mediated glucose metabolism. Cancer Cell Int. 2020, 20, 69.
- Wang, X.F.; Shi, Z.M.; Wang, X.R.; Cao, L.; Wang, Y.Y.; Zhang, J.X.; Yin, Y.; Luo, H.; Kang, C.S.; Liu, N.; et al. MiR-181d acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2. J. Cancer Res. Clin. Oncol. 2012, 138, 573–584.
- He, J.; Deng, Y.; Yang, G.; Xie, W. MicroRNA-203 down-regulation is associated with unfavorable prognosis in human glioma. J. Surg Oncol. 2013, 108, 121–125.
- Xiong, Z.; Zhou, C.; Wang, L.; Zhu, R.; Zhong, L.; Wan, D.; Wang, Q. Circular RNA SMO sponges miR-338-3p to promote the growth of glioma by enhancing the expression of SMO. Aging (Albany NY) 2019, 11, 12345–12360.
- Wu, X.; Hu, C.; Long, C.; Zhai, X.; Liang, P.; Yu, Z. MicroRNA-351 Promotes the Proliferation and Invasion of Glioma Cells through Downregulation of NAIF1. J. Mol. Neurosci. 2020, 70, 1493–1499.
- Ding, C.; Wu, Z.; You, H.; Ge, H.; Zheng, S.; Lin, Y.; Wu, X.; Lin, Z.; Kang, D. CircNFIX promotes progression of glioma through regulating miR-378e/RPN2 axis. J. Exp. Clin. Cancer Res. 2019, 38, 506.
- Li, S.; Zeng, A.; Hu, Q.; Yan, W.; Liu, Y.; You, Y. miR-423-5p contributes to a malignant phenotype and temozolomide chemoresistance in glioblastomas. Neuro Oncol. 2017, 19, 55–65.
- Zhang, X.; Wang, S.; Lin, G.; Wang, D. Down-regulation of circ-PTN suppresses cell proliferation, invasion and glycolysis in glioma by regulating miR-432-5p/RAB10 axis. Neurosci. Lett. 2020, 735, 135153.
- Nan, Y.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Yang, Y.; Yue, X.; Pu, P.; Zhong, Y.; Kang, C. MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res. 2010, 1359, 14–21.
- Liu, B.; Cao, W.; Ma, H. Knockdown of lncRNA LSINCT5 suppresses growth and metastasis of human glioma cells via up-regulating miR-451. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2507–2515.
- Shi, Y.; Chen, C.; Zhang, X.; Liu, Q.; Xu, J.L.; Zhang, H.R.; Yao, X.H.; Jiang, T.; He, Z.C.; Ren, Y.; et al. Primate-specific miR-663 functions as a tumor suppressor by targeting PIK3CD and predicts the prognosis of human glioblastoma. Clin. Cancer Res. 2014, 20, 1803–1813.
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773.
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498.
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744.
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30.
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469.
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567.
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307.
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265.
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488.
- Yao, J.; Chakhoyan, A.; Nathanson, D.A.; Yong, W.H.; Salamon, N.; Raymond, C.; Mareninov, S.; Lai, A.; Nghiemphu, P.L.; Prins, R.M.; et al. Metabolic characterization of human IDH mutant and wild type gliomas using simultaneous pH- and oxygen-sensitive molecular MRI. Neuro Oncol. 2019, 21, 1184–1196.
- Khurshed, M.; Molenaar, R.J.; Lenting, K.; Leenders, W.P.; van Noorden, C.J.F. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget 2017, 8, 49165–49177.
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Chaumeil, M.M.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS ONE 2015, 10, e0118781.
- Wenger, K.J.; Steinbach, J.P.; Bähr, O.; Pilatus, U.; Hattingen, E. Lower Lactate Levels and Lower Intracellular pH in Patients with IDH-Mutant versus Wild-Type Gliomas. AJNR Am. J. Neuroradiol. 2020, 41, 1414–1422.
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized (13)C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. Neuroimage Clin. 2016, 12, 180–189.
- Viswanath, P.; Najac, C.; Izquierdo-Garcia, J.L.; Pankov, A.; Hong, C.; Eriksson, P.; Costello, J.F.; Pieper, R.O.; Ronen, S.M. Mutant IDH1 expression is associated with down-regulation of monocarboxylate transporters. Oncotarget 2016, 7, 34942–34955.
- Ruiz-Rodado, V.; Malta, T.M.; Seki, T.; Lita, A.; Dowdy, T.; Celiku, O.; Cavazos-Saldana, A.; Li, A.; Liu, Y.; Han, S.; et al. Metabolic reprogramming associated with aggressiveness occurs in the G-CIMP-high molecular subtypes of IDH1mut lower grade gliomas. Neuro Oncol 2020, 22, 480–492.
- Karpel-Massler, G.; Ishida, C.T.; Bianchetti, E.; Zhang, Y.; Shu, C.; Tsujiuchi, T.; Banu, M.A.; Garcia, F.; Roth, K.A.; Bruce, J.N.; et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat. Commun. 2017, 8, 1067.
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515.
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700.
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446.
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell Biol. 2003, 23, 7315–7328.
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899.
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014.
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718.
- Ježek, P. 2-Hydroxyglutarate in Cancer Cells. Antioxid. Redox Signal. 2020, 33, 903–926.
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour. Biol. 2015, 36, 655–662.
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol. 2014, 127, 221–233.
- Mohrenz, I.V.; Antonietti, P.; Pusch, S.; Capper, D.; Balss, J.; Voigt, S.; Weissert, S.; Mukrowsky, A.; Frank, J.; Senft, C.; et al. Isocitrate dehydrogenase 1 mutant R132H sensitizes glioma cells to BCNU-induced oxidative stress and cell death. Apoptosis 2013, 18, 1416–1425.
- Bekaert, L.; Valable, S.; Lechapt-Zalcman, E.; Ponte, K.; Collet, S.; Constans, J.M.; Levallet, G.; Bordji, K.; Petit, E.; Branger, P.; et al. [18F]-FMISO PET study of hypoxia in gliomas before surgery: Correlation with molecular markers of hypoxia and angiogenesis. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1383–1392.
- Mapelli, P.; Callea, M.; Fallanca, F.; Castellano, A.; Bailo, M.; Scifo, P.; Bettinardi, V.; Conte, G.M.; Monterisi, C.; Rancoita, P.M.V.; et al. 18F-FAZA PET/CT in pretreatment assessment of hypoxic status in high-grade glioma: Correlation with hypoxia immunohistochemical biomarkers. Nucl. Med. Commun. 2021.
- Sadeghzadeh, M.; Wenzel, B.; Gündel, D.; Deuther-Conrad, W.; Toussaint, M.; Moldovan, R.P.; Fischer, S.; Ludwig, F.A.; Teodoro, R.; Jonnalagadda, S.; et al. Development of Novel Analogs of the Monocarboxylate Transporter Ligand FACH and Biological Validation of One Potential Radiotracer for Positron Emission Tomography (PET) Imaging. Molecules 2020, 25, 2309.
- Yang, X.; Zhu, H.; Li, N.; Huang, H.; Liu, T.; Guo, X.; Xu, X.; Xia, L.; Deng, C.; Tian, X.; et al. Targeting CAIX with [64 Cu]XYIMSR-06 Small Molecular Radiotracer Enables Noninvasive PET Imaging of Malignant Glioma in U87 MG Tumor Cell Xenograft Mice. Mol. Pharm. 2019, 16, 1532–1540.
- Radoul, M.; Najac, C.; Viswanath, P.; Mukherjee, J.; Kelly, M.; Gillespie, A.M.; Chaumeil, M.M.; Eriksson, P.; Delos Santos, R.; Pieper, R.O.; et al. HDAC inhibition in glioblastoma monitored by hyperpolarized 13C MRSI. Nmr. Biomed. 2019, 32, e4044.
- Lim, K.S.; Lim, K.J.; Price, A.C.; Orr, B.A.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2014, 33, 4433–4441.
- Takada, T.; Takata, K.; Ashihara, E. Inhibition of monocarboxylate transporter 1 suppresses the proliferation of glioblastoma stem cells. J. Physiol. Sci. 2016, 66, 387–396.
- Zhou, H.G.; Zhang, J.D.; Zhang, Y.F. [The effect of downregulation of MCT1 on the proliferation of glioma cells]. Zhonghua Zhong Liu Za Zhi 2019, 41, 208–213.
- Rafiki, A.; Boulland, J.L.; Halestrap, A.P.; Ottersen, O.P.; Bergersen, L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience 2003, 122, 677–688.
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823.
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: An in vivo study. Neoplasia 2011, 13, 620–632.
- Voss, D.M.; Spina, R.; Carter, D.L.; Lim, K.S.; Jeffery, C.J.; Bar, E.E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292.
- Miranda-Goncalves, V.; Cardoso-Carneiro, D.; Valbom, I.; Cury, F.P.; Silva, V.A.; Granja, S.; Reis, R.M.; Baltazar, F.; Martinho, O. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget 2017, 8, 103657–103670.
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.L.; Ledaki, I.; Turley, H.; Leek, R.; Snell, C.; Gatter, K.; Sly, W.S.; et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 2012, 18, 3100–3111.
- Proescholdt, M.A.; Merrill, M.J.; Stoerr, E.M.; Lohmeier, A.; Pohl, F.; Brawanski, A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro Oncol. 2012, 14, 1357–1366.
- Colen, C.B.; Seraji-Bozorgzad, N.; Marples, B.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: An in vitro study. Neurosurgery 2006, 59, 1313–1323.
- Amiri, A.; Le, P.U.; Moquin, A.; Machkalyan, G.; Petrecca, K.; Gillard, J.W.; Yoganathan, N.; Maysinger, D. Inhibition of carbonic anhydrase IX in glioblastoma multiforme. Eur. J. Pharm. Biopharm. 2016, 109, 81–92.
- Gillespie, D.L.; Aguirre, M.T.; Ravichandran, S.; Leishman, L.L.; Berrondo, C.; Gamboa, J.T.; Wang, L.; King, R.; Wang, X.; Tan, M.; et al. RNA interference targeting hypoxia-inducible factor 1α via a novel multifunctional surfactant attenuates glioma growth in an intracranial mouse model. J. Neurosurg. 2015, 122, 331–341.
- El Sayed, S.M.; El-Magd, R.M.; Shishido, Y.; Chung, S.P.; Diem, T.H.; Sakai, T.; Watanabe, H.; Kagami, S.; Fukui, K. 3-Bromopyruvate antagonizes effects of lactate and pyruvate, synergizes with citrate and exerts novel anti-glioma effects. J. Bioenerg. Biomembr. 2012, 44, 61–79.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Thiepold, A.L.; Lorenz, N.I.; Foltyn, M.; Engel, A.L.; Divé, I.; Urban, H.; Heller, S.; Bruns, I.; Hofmann, U.; Dröse, S.; et al. Mammalian target of rapamycin complex 1 activation sensitizes human glioma cells to hypoxia-induced cell death. Brain 2017, 140, 2623–2638.
- Xue, S.; Song, G.; Yu, J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Sci. Rep. 2017, 7, 4231.
- Givechian, K.B.; Garner, C.; Benz, S.; Rabizadeh, S.; Soon-Shiong, P. Glycolytic expression in lower-grade glioma reveals an epigenetic association between IDH mutation status and PDL1/2 expression. Neurooncol. Adv. 2021, 3, vdaa162.
- Vallejo, F.A.; Vanni, S.; Graham, R.M. UCP2 as a Potential Biomarker for Adjunctive Metabolic Therapies in Tumor Management. Front. Oncol. 2021, 11, 640720.
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643.
- O’Sullivan, D.; Pearce, E.L. Targeting T cell metabolism for therapy. Trends Immunol. 2015, 36, 71–80.
- Singer, K.; Kastenberger, M.; Gottfried, E.; Hammerschmied, C.G.; Büttner, M.; Aigner, M.; Seliger, B.; Walter, B.; Schlösser, H.; Hartmann, A.; et al. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int. J. Cancer 2011, 128, 2085–2095.
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241.
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 2010, 32, 67–78.
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488.
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Bristow, C.A.; Carugo, A.; Peoples, M.D.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674.
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256.
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819.
- Cui, J.; Zhang, Q.; Song, Q.; Wang, H.; Dmitriev, P.; Sun, M.Y.; Cao, X.; Wang, Y.; Guo, L.; Indig, I.H.; et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro. Oncol. 2019, 21, 1436–1446.
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436.
- Silva, L.S.; Poschet, G.; Nonnenmacher, Y.; Becker, H.M.; Sapcariu, S.; Gaupel, A.C.; Schlotter, M.; Wu, Y.; Kneisel, N.; Seiffert, M.; et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 2017, 18, 2172–2185.
- Huang, B.R.; Liu, Y.S.; Lai, S.W.; Lin, H.J.; Shen, C.K.; Yang, L.Y.; Lu, D.Y. CAIX Regulates GBM Motility and TAM Adhesion and Polarization through EGFR/STAT3 under Hypoxic Conditions. Int. J. Mol. Sci. 2020, 21, 5838.
- Lu, J.; Xu, Z.; Duan, H.; Ji, H.; Zhen, Z.; Li, B.; Wang, H.; Tang, H.; Zhou, J.; Guo, T.; et al. Tumor-associated macrophage interleukin-β promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci. 2020, 111, 1979–1990.
- Li, C.; Meng, G.; Su, L.; Chen, A.; Xia, M.; Xu, C.; Yu, D.; Jiang, A.; Wei, J. Dichloroacetate blocks aerobic glycolytic adaptation to attenuated measles virus and promotes viral replication leading to enhanced oncolysis in glioblastoma. Oncotarget 2015, 6, 1544–1555.

