 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Sol Jacome Burbano | + 1996 word(s) | 1996 | 2021-05-19 10:13:28 | | | |
| 2 | Karina Chen | Meta information modification | 1996 | 2021-06-02 09:04:41 | | |
Video Upload Options
To survive and reproduce, living organisms must maintain homeostasis both in unchallenged (normal) and challenged (stressful) contexts. This requires the evolution of powerful stress response mechanisms adapted to a particular ecosystem and to regular environmental fluctuations. Thus, these mechanisms may be very diverse within the tree of life. The pioneering work of Miroslav Radman on the stress response in bacteria demonstrated the rapid and adaptive value of changing mutation rates for rapid evolution (the mutator effect). In other words, to facilitate the survival of a species, whether it be to respond to a replication blockade or to a stressful environment, it is better to rapidly evolve by generating more mutations, some being possibly lethal, than to die immediately. We believe that this principle applies to the complex dynamics of telomeres in eukaryotes, which become altered in response to stress.
1. Telomeres: Basic Molecular Mechanisms in an Evolutionary Context
Telomeres are nucleoprotein structures at the ends of linear chromosomes that are required to maintain genome stability by providing a terminal cap [1]. Their function and replication require a complex interplay between specialized and general factors involved in replication and the DNA damage response. Strikingly, their structures change during an organism’s lifetime, both as a result of developmentally regulated programs and external factors. This, together with significant variability among organisms in telomere structure and regulation, raises several fundamental questions regarding telomere evolution and their contribution to development, life history, aging, disease susceptibility and the stress response. To understand fully the “telomere logic” in the stress response, it is necessary to understand the molecular challenges that these structures must face and the evolutionary solutions adopted by different organisms.
1.1. Challenge 1: The End-Stability Problem
In eukaryotes, the linearity of chromosomes requires the need to distinguish natural DNA termini from accidental breaks, to avoid unwanted recombination, karyotype instability and unwanted cell cycle arrest. The solution to this stability problem is the specific association of chromosome termini with protective factors including non-coding RNA (Telomeric Repeat-containing RNA or TERRA), and specialized proteins, for which the shelterin complex represents the paradigm [1].
In most eukaryotes, the telomeric DNA sequence is composed of G-rich DNA repeats ending at chromosome termini with a single-stranded DNA G-rich 3′-overhang (or G-tail) [2][3][4]. This 3′ overhang allows the formation of a telomeric DNA loop (t-loop) resulting from the invasion of the 3′ overhang into the internal duplex DNA. The formation of this t-loop is required to maintain telomere stability in a large range of organisms and in many different types of cells. Its formation is assisted by the shelterin subunit TRF2 [5].
The shelterin complex consists of proteins bound to the double-stranded part of telomeric DNA (e.g., TRF1 and TRF2 in chordates; Rap1 in Saccharomyces cerevisiae; DNT-1 and DNT-2 in Caenorhabditis elegans) [1][6] and single-stranded telomeric DNA (e.g., POT1 in humans; POT-1/POT-2 in nematodes). Finally, some shelterin subunits create a bridge between the duplex and the 3′ overhang nucleoprotein shelterin complexes (e.g., TPP1/ACD and TIN2 in mammals), or simply bind to specific other subunits (for example, mammalian RAP1 binds to TRF2) (Figure 1). One notable exception to this scheme is Drosophila melanogaster telomeric DNA, which is composed of long arrays of the non-long-terminal-repeat retrotransposons HeT-A, TART and TAHRE, bound by the Terminin protein complex that has no clear evolutionary relationship with shelterin and is elongated by retrotransposition, but also by recombination and gene conversion [7][8].
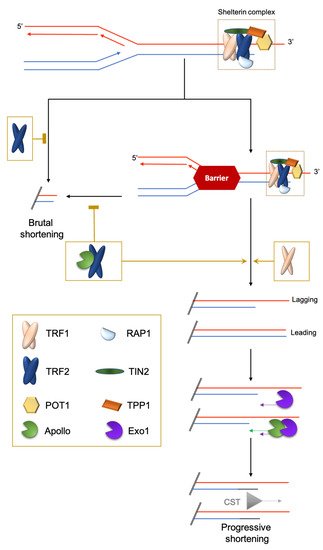
Figure 1. Brutal and progressive telomere DNA shortening. Telomere replication at the leading or lagging strands is not similarly regulated. At the G′-rich lagging strand, due to the end-replication problem, the parental strand is not completely replicated, triggering a 3′-overhang. At the leading strand, there is a blunt end or 5′protruding that is eroded in a manner coupled to replication by the TRF2-dependent recruitment of the 5′-exonuclease Apollo. Next, at both strands, the Exo1 exonuclease prolongs the 3′-overhang. Finally, POT1 recruits the CST complex (CTC1-STN1-TEN1) to replenish the 5′-strands. The brutal appearance of critically short telomeres that cannot be explained by the progressive telomere shortening was first observed in yeast [9]. They are likely to stem from intra-telomeric recombination [10][11] or the collapse of DNA replication forks. Indeed, it is known that telomeric repeats can form recombinogenic t-loops [12][13] and are difficult to replicate [14][15][16].
1.2. Challenge 2: End Replication Problem
There are several processes that compensate for this unavoidable shortening. One such mechanism relies on telomerase, a specialized reverse transcriptase with an RNA template that elongates the G-rich strand using the 3′ overhang as a substrate. Telomerase is composed of two subunits: TERT, the telomerase reverse transcriptase, and TERC, the telomerase RNA template. Using a single telomere length system and monitoring telomere DNA length (TL) evolution in budding yeast, it was shown that telomerase activity is reduced by over elongated telomeres: excess of Rap1p at the long yeast telomeres triggers a progressive cis-inhibition of telomerase [22]. This indicates that it is important to keep TL within a regulated range of size.
In mammalian species with large body mass, including humans and some birds, the level of telomerase expression is much higher in germ, stem and progenitor cells than in somatic differentiated cells. In some species, such as laboratory mice, telomerase expression is less repressed and more evenly distributed within cell types than in humans, but still higher in the germ and stem cell compartments [23][24][25]. Importantly, the amount of telomerase present in adult somatic stem cells and progenitors is generally not enough to replenish telomeric DNA fully when these cells divide to regenerate the corresponding tissue or to respond to external stimuli.
In many vertebrates, TL is subjected to a progressive erosion cadenced by cell divisions and the residual level of telomerase activity. There are also exceptions to this scheme; for instance, in the long-lived bat species Myotis myotis, mean TL does not shorten with age but changes with the environment [26]. In fact, the rate of telomere shortening in blood cells during development and aging is not strictly related to the level of telomerase activity [4][27]. This suggests that telomerase may have functions other than regulating telomere length and that TL might be regulated independently of telomerase. Indeed, the mean TL is the net result of shortening and elongation rates, rendering it difficult to interpret TL changes only in terms of telomerase-dependent elongation [22]. One interesting hypothesis is that the rate of telomere DNA shortening, but not the absolute length, controls the aging process [27]. It is not yet known whether there exist developmentally regulated mechanisms, independently of telomerase activity, that modulate the rate of TL shortening.
Interestingly, eukaryotic cells are able to use a telomerase-independent mechanism, ALT (alternative lengthening of telomeres), which is based on homologous recombination [28]. It is activated in a number of human tumors, in human cells immortalized in culture, and also in normal somatic tissues [29]. In plants, the ALT mechanism is activated upon telomerase dysfunction and possibly also during the earliest stages of normal plant development [30]. At the organismal level, ALT was suggested by Gomes and colleagues [4] for koala and wombat telomeres, based on the presence of long and interrupted telomere repeats and the absence of telomerase expression. However, it is not known whether these ALT-like telomeres use the same process of maintenance as the ALT + cancer cells. Along this line, a recent work that revealed the existence of an ALT-like mechanism in Terc knock-out (KO) mouse stem cells, with a unique non-telomeric sequence (mTALT) [31]. This process might be related to the one used in wombat. Hence, ALT or ALT-like mechanisms could give an advantage in the context of species with no telomerase. In order to avoid cancer development, we can imagine ALT process is finely regulated in species using only this pathway to preserve their telomeres.
1.3. Challenge 3: The Hard-to-Replicate Problem
In addition to the end-replication problem, the progression of the replication fork through telomeric chromatin is difficult and may result in fork collapses leading to the loss of a large part of the telomeric DNA. This is prevented by specific mechanisms involving the shelterin subunits TRF1 and TRF2: TRF1 manages classical stalled forks leading to ATR activation and TRF2 manages topologically-constrained stalled forks [16][32] (Figure 1).
Interestingly, the function of TRF2 in this process requires the same factor, Apollo, as the one favoring the progressive shortening of the leading strand [16]. Inhibiting TRF2-Apollo interaction is associated with severe progeroid syndrome in humans [33]. The long-lived giant tortoises Chelonoidis abingdonii and Aldabrachelys gigantea carry an Apollo variation within the TRF2 binding domain that is not conserved in other reptiles, fishes or mammals and, thus, could contribute to the extreme longevity of these organisms [34]. Altogether, these findings suggest an evolutionary advantage, at least in some organisms, to control progressive shortening due to the end replication problem. This hypothesis seems at odds with the wealth of studies showing that TL is positively associated with improved healthspan and lifespan in vertebrates [35][36]. Usually, short telomeres are associated with chronological aging, cardiovascular and age-related diseases and poor prognosis [37]. However, long telomeres are not as beneficial as was initially thought: several studies have shown an association between long telomeres and some types of cancer. Actually, in a genome-wide association study, nine cancers out of 35 (including glioma, neuroblastoma and endometrial cancer) were associated with increased TL [38]. To understand the “raison d’être” of these different telomere trajectories between organisms and individuals, and their consequences for health, we need to understand more fully the complex interplay between genetic and environmental determinants regulating the response of telomeres to stress during development and aging.
2. Telomere Response to Stress
A stressor can be defined as a factor that leads to a rupture in homeostasis, whether it be at the cellular, systemic or organismal level. Thus, the environment can be defined as the combination of stressful situations to which individuals are exposed during their lifetime, including lifestyle, climate, pollutants, pathogens and social relationships. Organisms have evolved mechanisms to cope with their environments and predictable environmental fluctuations. Abnormal exposure to stressors is a well-known risk of age-related disease and premature aging [39]. Different types of stressors also lead to an acceleration of TL shortening, leading to critically short telomeres and cellular senescence, a mechanism that could explain the link between stress and aging. To give an example, the telomere shortening rate can increase by roughly five times in fibroblast cultured in hyperoxia compared to normoxia [40]. The rapidity by which these changes are signaled to the cells is not well understood. One can imagine two non-mutually exclusive modes of signaling. On one hand, shortened telomeres are phenotypically “silent” until they reach a critically short length that triggers senescence. On the other hand, even small changes in telomere length variation can have an impact, even before cells reach senescence. In the following paragraphs, we discuss how various stresses can modulate telomere structure and function (Figure 2).
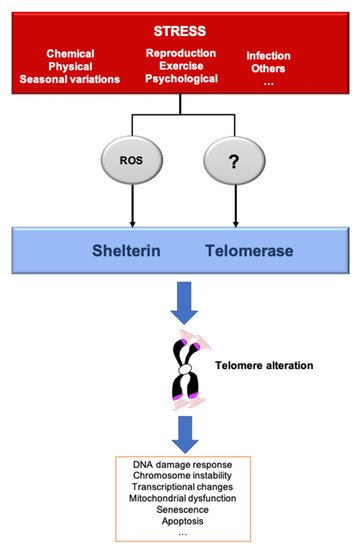
Figure 2. Stressors affect telomere homeostasis. Several stresses such as changes in pH or temperature, exercise, irradiation, pathogens infection, reproduction/breeding efforts, exposure to chemicals or psychological pressure, among others, can affect telomere integrity. The most described mechanisms induced by several of these stressors include telomerase inhibition and an increase of ROS, that preferentially oxidizes the telomeric DNA, a process that we dubbed teloxidation [41]. Upon challenge, telomeric alterations induce the DNA damage response activation (DDR), senescence, transcriptional changes and mitochondrial dysfunction.
References
- Giraud-Panis, M.-J.; Pisano, S.; Poulet, A.; Le Du, M.-H.; Gilson, E. Structural Identity of Telomeric Complexes. FEBS Lett. 2010, 584.
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A Highly Conserved Repetitive DNA Sequence, (TTAGGG)n, Present at the Telomeres of Human Chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626.
- Meyne, J.; Ratliff, R.L.; Moyzis, R.K. Conservation of the Human Telomere Sequence (TTAGGG)n among Vertebrates. Proc. Natl. Acad. Sci. USA 1989, 86, 7049–7053.
- Gomes, N.M.V.; Ryder, O.A.; Houck, M.L.; Charter, S.J.; Walker, W.; Forsyth, N.R.; Austad, S.N.; Venditti, C.; Pagel, M.; Shay, J.W.; et al. Comparative Biology of Mammalian Telomeres: Hypotheses on Ancestral States and the Roles of Telomeres in Longevity Determination. Aging Cell 2011, 10, 761–768.
- Benarroch-Popivker, D.; Pisano, S.; Mendez-Bermudez, A.; Lototska, L.; Kaur, P.; Bauwens, S.; Djerbi, N.; Latrick, C.M.; Fraisier, V.; Pei, B.; et al. TRF2-Mediated Control of Telomere DNA Topology as a Mechanism for Chromosome-End Protection. Mol. Cell 2016, 61, 274–286.
- Yamamoto, I.; Zhang, K.; Zhang, J.; Vorontsov, E.; Shibuya, H. Telomeric Double-Strand DNA-Binding Proteins DTN-1 and DTN-2 Ensure Germline Immortality in Caenorhabditis Elegans. eLife 2021, 10, e64104.
- Mason, J.M.; Frydrychova, R.C.; Biessmann, H. Drosophila Telomeres: An Exception Providing New Insights. BioEssays 2008, 30, 25–37.
- Cacchione, S.; Cenci, G.; Raffa, G.D. Silence at the End: How Drosophila Regulates Expression and Transposition of Telomeric Retroelements. J. Mol. Biol. 2020, 432, 4305–4321.
- Chang, M.; Arneric, M.; Lingner, J. Telomerase Repeat Addition Processivity Is Increased at Critically Short Telomeres in a Tel1-Dependent Manner in Saccharomyces Cerevisiae. Genes Dev. 2007, 21, 2485–2494.
- Ancelin, K.; Brunori, M.; Bauwens, S.; Koering, C.-E.; Brun, C.; Ricoul, M.; Pommier, J.-P.; Sabatier, L.; Gilson, E. Targeting Assay To Study the Cis Functions of Human Telomeric Proteins: Evidence for Inhibition of Telomerase by TRF1 and for Activation of Telomere Degradation by TRF2. Mol. Cell. Biol. 2002, 22, 3474–3487.
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of Telomere Length by a Trimming Mechanism That Involves Generation of T-Circles. EMBO J. 2009, 28, 799–809.
- Wang, R.C.; Smogorzewska, A.; de Lange, T. Homologous Recombination Generates T-Loop-Sized Deletions at Human Telomeres. Cell 2004, 119, 355–368.
- Saint-Léger, A.; Koelblen, M.; Civitelli, L.; Bah, A.; Djerbi, N.; Giraud-Panis, M.-J.; Londoño-Vallejo, A.; Ascenzioni, F.; Gilson, E. The Basic N-Terminal Domain of TRF2 Limits Recombination Endonuclease Action at Human Telomeres. Cell Cycle 2014, 13, 2469–2474.
- Miller, K.M.; Rog, O.; Cooper, J.P. Semi-Conservative DNA Replication through Telomeres Requires Taz1. Nature 2006, 440, 824–828.
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. Cell 2009, 138, 90–103.
- Ye, J.; Lenain, C.; Bauwens, S.; Rizzo, A.; Saint-Léger, A.; Poulet, A.; Benarroch, D.; Magdinier, F.; Morere, J.; Amiard, S.; et al. TRF2 and Apollo Cooperate with Topoisomerase 2alpha to Protect Human Telomeres from Replicative Damage. Cell 2010, 142, 230–242.
- Watson, J.D. Origin of Concatemeric T7DNA. Nat. New Biol. 1972, 239, 197–201.
- Olovnikov, A.M. A Theory of Marginotomy. The Incomplete Copying of Template Margin in Enzymic Synthesis of Polynucleotides and Biological Significance of the Phenomenon. J. Theor. Biol. 1973, 41, 181–190.
- Lingner, J.; Cooper, J.P.; Cech, T.R. Telomerase and DNA End Replication: No Longer a Lagging Strand Problem? Science 1995, 269, 1533–1534.
- Gilson, E.; Géli, V. How Telomeres Are Replicated. Nat. Rev. Mol. Cell. Biol. 2007, 8, 825–838.
- Wu, P.; Takai, H.; de Lange, T. Telomeric 3′ Overhangs Derive from Resection by Exo1 and Apollo and Fill-in by POT1b-Associated CST. Cell 2012, 150, 39–52.
- Marcand, S.; Brevet, V.; Gilson, E. Progressive Cis-Inhibition of Telomerase upon Telomere Elongation. EMBO J. 1999, 18, 3509–3519.
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase Activity in Human Germline and Embryonic Tissues and Cells. Dev. Genet. 1996, 173–179.
- Haussmann, M.F.; Winkler, D.W.; Huntington, C.E.; Nisbet, I.C.T.; Vleck, C.M. Telomerase Activity Is Maintained throughout the Lifespan of Long-Lived Birds. Exp. Gerontol. 2007, 42, 610–618.
- Roake, C.M.; Artandi, S.E. Regulation of Human Telomerase in Homeostasis and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397.
- Foley, N.M.; Hughes, G.M.; Huang, Z.; Clarke, M.; Jebb, D.; Whelan, C.V.; Petit, E.J.; Touzalin, F.; Farcy, O.; Jones, G.; et al. Growing Old, yet Staying Young: The Role of Telomeres in Bats’ Exceptional Longevity. Sci. Adv. 2018, 4, eaao0926.
- Whittemore, K.; Vera, E.; Martínez-Nevado, E.; Sanpera, C.; Blasco, M.A. Telomere Shortening Rate Predicts Species Life Span. Proc. Natl. Acad. Sci. USA 2019, 116, 15122–15127.
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an Alternative Mechanism for Maintaining Telomere Length in Human Tumors and Tumor-Derived Cell Lines. Nat. Med. 1997, 3, 1271–1274.
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.L.; Reddel, R.R. Alternative Lengthening of Telomeres in Normal Mammalian Somatic Cells. Genes Dev. 2013, 27, 18–23.
- Role of Alternative Telomere Lengthening Unmasked in Telomerase Knock-Out Mutant Plants | SpringerLink. Available online: (accessed on 28 April 2021).
- Kim, C.; Sung, S.; Kim, J.-S.; Lee, H.; Jung, Y.; Shin, S.; Kim, E.; Seo, J.J.; Kim, J.; Kim, D.; et al. Telomeres Reforged with Non-Telomeric Sequences in Mouse Embryonic Stem Cells. Nat. Commun. 2021, 12, 1097.
- Ohki, R.; Ishikawa, F. Telomere-Bound TRF1 and TRF2 Stall the Replication Fork at Telomeric Repeats. Nucleic Acids Res. 2004, 32, 1627–1637.
- Touzot, F.; Callebaut, I.; Soulier, J.; Gaillard, L.; Azerrad, C.; Durandy, A.; Fischer, A.; de Villartay, J.-P.; Revy, P. Function of Apollo (SNM1B) at Telomere Highlighted by a Splice Variant Identified in a Patient with Hoyeraal–Hreidarsson Syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10097–10102.
- Quesada, V.; Freitas-Rodríguez, S.; Miller, J.; Pérez-Silva, J.G.; Jiang, Z.-F.; Tapia, W.; Santiago-Fernández, O.; Campos-Iglesias, D.; Kuderna, L.F.K.; Quinzin, M.; et al. Giant Tortoise Genomes Provide Insights into Longevity and Age-Related Disease. Nat. Ecol. Evol. 2019, 3, 87–95.
- Aviv, A. Telomeres and Human Somatic Fitness. J. Gerontol. Ser. A 2006, 61, 871–873.
- Heidinger, B.J.; Blount, J.D.; Boner, W.; Griffiths, K.; Metcalfe, N.B.; Monaghan, P. Telomere Length in Early Life Predicts Lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 1743–1748.
- Aviv, A.; Shay, J.W. Reflections on Telomere Dynamics and Ageing-Related Diseases in Humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373.
- The Telomeres Mendelian Randomization Collaboration; Haycock, P.C.; Burgess, S.; Nounu, A.; Zheng, J.; Okoli, G.N.; Bowden, J.; Wade, K.H.; Timpson, N.J.; Evans, D.M.; et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 2017, 3, 636.
- de Magalhães, J.P.; Passos, J.F. Stress, Cell Senescence and Organismal Ageing. Mech. Ageing Dev. 2018, 170, 2–9.
- von Zglinicki, T.; Saretzki, G.; Döcke, W.; Lotze, C. Mild Hyperoxia Shortens Telomeres and Inhibits Proliferation of Fibroblasts: A Model for Senescence? Exp. Cell Res. 1995, 220, 186–193.
- Jacome Burbano, M.S.; Cherfils-Vicini, J.; Gilson, E. Neutrophils: Mediating TelOxidation and Senescence. EMBO J. 2021, 40, e108164.



