 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marcello Persico | + 3848 word(s) | 3848 | 2021-05-19 10:58:19 | | | |
| 2 | Karina Chen | Meta information modification | 3848 | 2021-06-01 04:59:04 | | |
Video Upload Options
Metabolic- (dysfunction) associated fatty liver disease (MAFLD) represents the predominant hepatopathy and one of the most important systemic, metabolic-related disorders all over the world associated with severe medical and socio-economic repercussions due to its growing prevalence, clinical course (steatohepatitis and/or hepatocellular-carcinoma), and related extra-hepatic comorbidities.
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) represents one of the most important metabolic-related disorders of the 21st century and the leading cause of chronic liver disease and liver transplantation worldwide [1]. It includes a wide spectrum of conditions ranging from simple steatosis to steatohepatitis (NASH), characterized by the histologic appearance of inflammation and fibrosis, which act as driving factors to fuel the disease progression and complications onset [2].
Currently, NAFLD is considered the hepatic manifestation of metabolic syndrome (MS) [3] and recently, an expert consensus established that metabolic- (dysfunction) associated fatty liver disease (MAFLD) represents the more adequate denomination, revealing its larger and deeper nature as systemic disorder [4] (Figure 1).
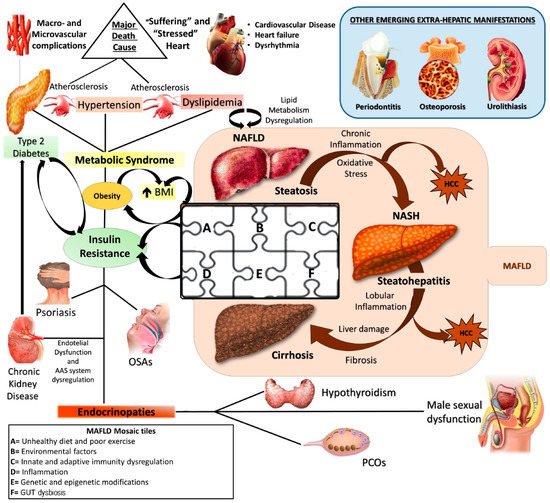
Figure 1. NAFLD: a metabolic systemic disease. The modern approach considers NAFLD as a metabolic, systemic disease characterized by several extra-hepatic manifestations mostly linked by a status of insulin resistance (IR). EDCs: Endocrine disrupting compounds; IR: Insulin resistance; MAFLD: metabolic (dysfunction) associated fatty liver disease; NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis; HCC: Hepatocellular carcinoma; OSA: Obstructive sleep apnea; PCOS: Polycystic ovarian syndrome; T2DM: Type 2 Diabetes Mellitus.
Alarmingly, as the diffusion of MS-related conditions continues to increase and the obesity pandemic spreads irrepressibly, MAFLD prevalence, in turn, seems to rise exponentially [1]. The worldwide prevalence ranges from 6% to 35%, with higher levels in the industrialized countries (the Middle East 32%, South America 31%, United States 24%, and Europe 23%) and lower levels in the underdeveloped ones (14%) [5]. Moreover, future perspectives appear decidedly not encouraging, expecting prevalence to reach about 100 million in the United States alone by 2030 [1].
The numbers of the MAFLD spread worldwide raise several important concerns principally due to the very complex and still not fully understood pathogenesis. It gives to the disease an inherent difficulty to support the burden of its optimal medical and social management.
Pathogenetically MAFLD represents a multifactorial disease in which various elements can simultaneously contribute to the genesis and affect natural history, contributing to make its manifestation from patient to patient hugely different [6]. The genome-wide association studies (GWAS) shed light on the genetic susceptibility of the population to MAFLD onset and evolution, identifying several single nucleotide polymorphisms (SNPs), like phospholipase domain-containing protein-3 (PNPLA3) rs738409, the transmembrane 6 superfamily member 2 protein (TM6SF2) rs58542926, and membrane-bound O-acyltransferase domain containing 7 (MBOAT7) rs641738, as crucial in this scenario [7][8]. At the same time, the contemporary involvement of several environmental factors like sedentary life, unhealthy diet regimens promoting insulin resistance (IR), and gut dysbiosis, together with some immune disturbances, have been accepted by the scientific community as “tiles of MAFLD pathogenetic mosaic” [9][10][11][12] (Figure 1).
To date, no specific medications for MAFLD treatment exist [5][13]; hence, given its demonstrated capability to induce huge improvement in IR and liver damage, lifestyle modification remains the most valid and accepted recommendation [14]. This approach classically rests on three enchained pivots: regular physical exercise, weight loss, and healthy diet [15]. Regarding the latter, the scientific evidence suggests the possibility that a regimen characterized by the main consumption of plant-based food, fish, and white meat could contribute significantly to the reduction of several chronic diseases’ occurrence, including MS components and thus MAFLD [16][17][18]. These principles constitute the paradigm of the well-known Mediterranean diet (MD), which, based on the antioxidant and anti-inflammatory properties of the recommended food, currently represents the nutritional gold standard of preventive medicine [19].
However, the different kinds of therapeutic outcomes potentially obtained from this dietary regimen induced the scientific community to focus attention on the identification of the factors involved in determining the dietary therapeutic effect, hypothesizing a mutual relationship between diet and genetics [15]. Recent findings suggested nutrition’s capability, by acting on the individual genetic background and modifying the specific epigenetic expression as well, to influence NAFLD patients’ clinical severity and response to the treatment [20]. The study of the interaction between nutrients and inherited factors constitutes the central aim of emerging and promising lines of research known as nutrigenetics and nutrigenomics [21].
Globally, the main scope of these research fields is strictly linked in a very complex scientific network with other disciplines, such as genomics, transcriptomics, proteomics, metabolomics, and system biology, configuring the “Omics-approach” to the disease that seems to be the key for the full comprehension of MAFLD pathogenesis and evolution [21][22] (Figure 2).
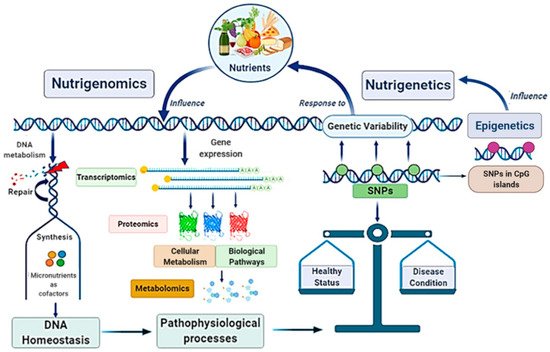
Figure 2. Nutrigenomics and nutrigenetics: two sides of the same medal with several linked approaches. Genome influences the responsiveness to nutrients (nutrigenetics’ field); at the same time, nutrition can also modify gene expression involving epigenetic mechanisms (nutrigenomics’ field). Nutrigenomics investigates the effects deriving from the interaction between the nutritional environment and inherited factors. Given the complexity of the scenario, nutrigenomics quests include several approaches involving many other disciplines. Nutritional factors and genetic ones influence each other: on one side, nutrients affect DNA metabolism, gene expression, and genetic variability; on the other side, genetic variants (as SNPs), by determining specific individual genotype, influence dietary habits. In turn, nutrigenetics could also be influenced by epigenetics. Altogether, these mechanisms contribute to determinate the status of health or a condition disease. SNPs, Single Nucleotide Polymorphisms.
2. Genetics and Epigenetics: An Overview on MAFLD Genetic Background
In MAFLD context, heritability seems to play a pivotal role as suggested clearly just considering the higher disease risk of offspring in case of positive family history for MAFLD, particularly when both parents are affected [23][24]. Furthermore, the ethnic susceptibility represents another overwhelming proof. Different ethnicities, in fact, show different propensity towards disease development and progression [25][26]. Interestingly, besides the inter-ethnic one, growing evidence reveals also an inter-individual variability. Therefore, people belonging to the same ethnicity have different possibilities to develop MAFLD, and this appears strictly linked to certain genetic variants as well as genetic expressions able to influence its onset and evolution [27][28][29].
2.1. Most Common Genetic Determinants of MAFLD
A long list of genetic inter-individual variants has been provided by GWAS in this context; it embraces several genes known as genetic determinants of MAFLD, whose expression is involved in the formation of lipid droplets and regulation of oxidative stress, inflammation, fibrogenesis, and many other metabolic pathways [7][29][30] (Table 1).
Table 1. Main genetic determinants of NAFLD.
| Gene | Variants/SNPs/Protein Variants | Relative Effects/Association with | |
|---|---|---|---|
| Major and most common genetic determinants of MAFLD | PNPLA3 | rs738409 (I148M) |
Disruption of triglycerides and phospholipids turnover and remodelling: increased hepatic fat accumulation; Disruption of retinol storage in HSCs leading to higher risk of inflammation, fibrosis, and HCC progression. |
| MBOAT 7 | rs641738 | Higher risk of MAFLD development, inflammation, fibrosis, and HCC progression. | |
| TM6SF2 | rs58542926 | Favouring liver fat accumulation;Protection against the development of cardiovascular diseases. | |
| Other genetic determinants involved in lipid metabolism | GCKR | rs1260326 | Increased de novo lipogenesis and worsened hepatic steatosis. |
| PPP1R3B | rs4841132 | Reduction of de novo lipogenesis and thus protection from hepatic fat accumulation. | |
| APOB | Several and different | Reduced VLDL export from hepatocytes. | |
| Other genetic determinants involved in oxidative stress imbalance | SOD2 | rs4880 | Higher oxidative stress and more advanced fibrosis. |
| UCP3 | rs1800849 | IR worsening, increased adiponectin levels, and NASH development. | |
| UCP2 | rs695366 | Higher insulin sensitivity and protection against liver damage. | |
| MARC1 | A165T | Lower hepatic fat accumulation and decreased levels of several biomarkers of liver disease. | |
| HFE | rs1800562 (C282Y) | Iron overload and related oxidative stress imbalance. | |
| Other genetic determinants involved in inflammation and fibrosis | TLR4 | D299G and T399I | Protection against fibrosis (in animal models). |
| IFNL4 | rs368234815 | Induction of severe inflammation. | |
| IFN/IL-28 | rs12979860 | Promotes inflammation and fibrosis (it is predictive for advanced stage of the disease). | |
| PCSK7 | rs236918 | Liver damage and altered fibrogenesis association. | |
| MERTK | rs4374383 | Protection against fibrosis. | |
| HSD17B13 | rs72613567 | Reduced risk of NASH (but not steatosis). |
However, more robust and reproducible associations seem to exist, above all, for PNPLA3, MBOAT7, and TM6SF2 variants that currently are considered the main genetic MAFLD determinants [8].
The rs738409 SNP of the PNPLA3 gene (or Adiponutrin), encoding for the I148M protein variant, showed heavy impact on disease susceptibility. This variant has been associated with higher hepatic fat content in an IR-independent manner, as demonstrated by Romeo et al. [31]. In details, the role of IR in the light of the current scientific knowledge regarding this purpose seems to be extremely complex and, from a certain point of view, still not fully understood. In fact, the PNPLA3 gene encodes for the homonymous transmembrane protein localized on the endoplasmic reticulum [32], mainly expressed in hepatocytes and adipose tissue and exhibiting triglycerides (TG) hydrolase activity regulated by glucose and insulin [32][33]. In case of IR and obesity, both conditions featured by high insulin levels, PNPLA3 expression is induced, and the protein is placed and accumulated on the surface of lipid droplets where it is not catalytically active, disrupting TG and phospholipids turnover and remodelling [34].
PNPLA3 is also involved in the release of the storage form of retinol, known as retinyl-palmitate, in hepatic stellate cells (HSCs) [35][36]. Due to the involvement of retinyl-palmitate in the regulation of cellular fat metabolism and HSCs activation, its retention caused by the I148M genetic variant determines, consequently, the activation of pro-inflammatory and pro-fibrotic responses [35][37]. About that, growing evidence in animal and clinical experimental models suggests the I148M genetic variant relationship with all the steps of the diseases’ natural history, from simple steatosis to NASH, cirrhosis, and hepatocellular carcinoma (HCC) development [38][39]. Moreover, this SNP appears to be involved as the key driver in chronic kidney disease (CKD) development and is currently recognized as an established marker of higher cardiovascular risk in MAFLD [40].
MBOAT7 is a gene encoding for a membrane-bound enzyme whose main function is the incorporation of arachidonic acid (AA) and other unsaturated fatty acids (UFAs) in the phosphatidylinositol (PI) molecule by the Lands cycle (a series of phospholipid-remodelling reactions by which acyl-chains become trans acylated) [41]. In the presence of the rs641738 variant, the expression of the encoded enzyme decreases together with the hepatic levels of PI containing AA and causes a disruption of several cellular pathways regulating TG metabolism, inflammation, fibrosis, and cellular proliferation as well [42][43][44].
The TM6SF2 gene is responsible for the production of a protein mainly localized in the endoplasmic reticulum and Golgi apparatus, involved in the regulation of the hepatic triglyceride secretion [45][46]. The rs58542926 C>T encoding for the E167K variant of this gene is involved in MAFLD development and worsening of histological picture, impairing inflammation, ballooning, and fibrosis, as demonstrated by Liu et al. [42][47]. On the other side, this SNP seems to confer protection from the cardiovascular disease risk through the reduction of lipid secretion and very-low-density lipoproteins (VLDL) synthesis, designing a very complex biologic role that still remains to be fully clarified [48][49][50].
Besides PNPLA3, MBOAT7, and TM6SF2 SNPs, other genetic variants appear potentially involved in this scenario. Strong scientific evidence exists for the glucokinase regulator (GCKR) common missense variants rs1260326 encoding for the P446L protein [51]. The common missense variant rs1260326 disrupts GCKR function, making it unable to inhibit the glucokinase and, consequently, activating hepatic glucose uptake and glycolysis. These phenomena lead to the generation of acetyl-CoA cellular overload and de novo lipogenesis (DNL) [51]. Contrariwise, the protein phosphatase 1 regulatory subunit 3B (PPP1R3B) rs4841132 variant, through the reduction of lipogenesis and the increase of glycogen synthesis, seems to protect against hepatic fat accumulation [52]. Besides the aforementioned one involved in steatosis development, other genetic polymorphisms have been associated with the disease progression.
As known, reactive oxygen species (ROS) overproduction derived from free fatty acids (FFAs) overload in mitochondria secondary to IR and consequent mitochondrial dysfunction, represent critical events in MAFLD worsening [53]. In this regard, some studies highlighted the association of polymorphism rs4880 of Superoxide dismutase 2 (SOD 2) with higher oxidative stress levels, inflammation, and more advanced fibrosis [54][55]. In line with this, the uncoupling protein 3 (UCP3) rs1800849, uncoupling protein 2 (UCP2) rs695366, homeostatic iron regulator (HFE) C282Y rs1800562, and a rare missense variant (A165T) in mitochondrial amidoxime reducing component 1 (MARC1) represent other new interesting topics [7][56][57]. UCP3 is a mitochondrial anion carrier selectively expressed in skeletal muscle involved in the modulation of energy, lipid homeostasis, and thermogenesis by facilitating the proton leak of the mitochondrial inner membrane and uncoupling the oxidative phosphorylation [58]. Interestingly, a Spanish study conducted on a cohort of overweight patients revealed the association of UCP3 rs1800849 variant with IR, increased adiponectin levels, and NASH [59]. On the contrary, the presence of the UCP2 rs695366 variant has been linked to higher gene expression, insulin sensitivity, and protection from liver damage [60].
Other rare genetic variants were demonstrated related to MAFLD clinical context as mainly involved in the disease progression instead of its development: the rs236918 genetic variant in proprotein convertase subtilisin/Kexin type 7 (PCSK7) [61], or protecting from the disease and complication onset: the loss of function in HSD17B13 gene due to rs72613567 variant [62][63].
However, one of the most intriguing questions is how far we are to apply in the routine clinical practice the knowledge derived from GWAS studies. This question could be interpreted from some different point of views on MAFLD clinical picture: predictive, prognostic, and therapeutic ones [28]. On this line, it is necessary to highlight the lack of scientific agreement regarding the best methodological choice to evaluate the utility of genetic variant risk estimates [64]. Considering the PNPLA3 I148M variant, it has great power in the prediction of the disease, which was highlighted by several clinical trials in which its role in disease appearance was totally demonstrated independent from the other classical risk factors [24][65]. However, its pertinence as a heritable factor for NAFLD development was not demonstrated as brilliant as the latter purpose and, for this reason, the actual clinical management guidelines of the European Association for the Study of the Liver do not recommend its routine assessment for NAFLD-related liver damage evaluation [66]. In this regard, because of the lack of sufficient scientific proof to support the use of a single SNP in the prediction of the disease risk, the recent scientific literature has focused attention on the possible applicability of polygenic risk scores (PRSs) for this purpose [64]. This acquires even more relevance considering the accuracy of PRS based on well-established SNP and commonly recognized risk factors for the disease development in NAFLD risk prediction [67][68]. The use of PRS in a cross-sectional study on NAFLD was demonstrated able to induce an improvement of risk prediction in about 20% of patients. Moreover, recently, PNPLA3-TM6SF2-GCKR-MBOAT7 variants combined in a hepatic fat PRS (PRS-HFC) and adjusted for HSD17B13 (PRS-5) were demonstrated as able to predict HCC more efficiently than single variants assessment and that the association between PRS and HCC, mediated by severe fibrosis, was independent from the latter in clinically relevant subgroups and in those without advanced stages fibrosis [69]. Despite that the emerging research topic could have huge scientific impact on future clinical management, the amount of scientific evidence currently remains the most important concern to recommend their routine use. Moreover, regarding the prediction of long-term outcome, independently from the baseline staging of the disease, nowadays, it is not possible to state scientifically coherent conclusions, at least until the publication of data from long-term prospective studies.
The possibility of MAFLD therapeutic outcome prediction in the era of the patients-tailored approach assumes a fascinating aura in the light of some research trials on new therapeutic agents for disease treatment [70]. This novel research line could give the possibility to interpret better the usefulness of a wide range of already known drugs and newly developed ones as well. The WELCOME trial evaluated the response to 4 × 1000-mg capsules of 460 mg eicosapentaenoic acid and 380 mg docosahexaenoic acid administration for 15–18 months on liver fat content and fibrosis in NAFLD patients [71]. At the end of treatment, those patients carrying the 148I/I and 148I/M genotype showed a decrease of liver fat percentage (148I/I: −7.05%, 148I/M: −7.30%) whereas the 148M/M group showed a moderate increase (2.75%) [71].
Even if for some of the identified genes more powerful scientific evidences are still missing and the results of the different studies appear sometimes controversial as well, there is no doubt in considering this field one of the most promising topics of the recent MAFLD research.
2.2. Main Epigenetic Mechanisms of MAFLD
As part of the complex genetic background sustaining MAFLD, several epigenetic phenomena, influencing different levels of gene expression regulation, seem to be potentially involved in pathogenesis and clinical history. The mechanisms described encompass DNA methylation, histone modifications, and microRNAs (miRNAs) activity on specific targets [72]. In this context, the development of a characteristic methylation pattern could be critical and fuel the disease progression [73]. Kitamoto et al. compared the levels of DNA methylation of certain CpG islands as CpG99 (which resides in the regulatory region of PNPLA3) and CpG26 (which resides in that of PARVB variant 1) in the livers of patients with mild (fibrosis stages 0 and 1) or advanced (fibrosis stages 2 to 4) steatosis by performing targeted-bisulfite sequencing [74]. Relevantly, in the livers of patients with advanced disease, CpG26 resulted markedly hypomethylated while CpG99 was substantially hypermethylated, suggesting the hypomethylation of CpG26 and the hypermethylation of CpG99 as potential contributors to the severity of fibrosis in patients with MAFLD [74]. Moreover, in individuals affected by severe steatosis compared to mild ones, lower DNA methylation levels characterized specific CpG islands of noted pro-fibrotic genes, such as transforming growth factor- β(TGF-β), collagen 1A1, platelet-derived growth factor- α(PDGF-α), and others [75][76]. On the contrary, hypermethylation status occurred for certain CpG islands in various anti-steatotic and anti-fibrotic genes, such as ApoB and peroxisome proliferator-activated receptor (PPAR)-α [75][77][78]. The loss of PPAR-α functioning seems extremely important because it regulates cytokine production, reducing the expression of pro-inflammatory ones, and, on the other hand, it modulates the proteins involved in the fatty acid binding activity, taking part in the regulation of lipogenesis during the oxidation [77][79].
In this sense, a large number of liver DNA methylation alterations widely affecting disease onset and worsening by promoting inflammation and fibrosis has been related to certain metabolic features in terms of insulin, amino acids, and lipids serum levels. An interesting analysis of the DNA methylation pattern performed on the liver biopsies obtained from 95 obese individuals (34 individuals showing normal liver phenotype, 35 simple steatosis, and 26 steatohepatitis), identified 1292 CpG sites representing 677 unique genes differentially methylated in the livers of individuals with advanced disease (i.e., steatohepatitis) [80]. Focusing on the top-ranking 30 and another 37 CpG sites mapped to genes enriched in pathways of metabolism and cancer all together, the authors revealed 59 steatohepatitis-associated CpG sites correlating with fasting insulin levels independently of age, fasting glucose, or diabetes mellitus type 2 [80].
In addition, in a recent study on 194 obese patients (79 with liver histology classifiable as normal liver, 40 as simple steatosis, and 45 as steatohepatitis) aiming to assess serum aromatic and branched-chain amino acids levels association with steatohepatitis, the tryptophan resulted significantly higher in those with advanced disease compared to those with simple steatosis [81]. Relevantly, these amino acid serum levels result were associated with liver DNA methylation of CpG sites known to be differentially methylated in individuals with steatohepatitis and were correlated positively with serum total and low-density lipoprotein (LDL) cholesterol and, accordingly, with liver low-density lipoprotein receptor (LDL-R) at mRNA-expression level [81].
Histone modifications represent another epigenetic phenomenon potentially related to different metabolic dysfunctions [72]. The disruption of circadian rhythm of histone acetylation regulated by histone deacetylases (HDCA3), implicated in the regulation of the circadian rhythm of hepatic lipogenesis [82], may alter hepatic lipid metabolism leading to IR and obesity [83][84]. In particular, a group of deacetylates, known as silent information regulator 2 proteins (Sirtuins), may exert a key role in MAFLD development [84], according to growing evidence revealing their downregulation in animals and in vitro models [84][85]. In mice, in fact, liver-specific deletion of SIRT1 by impairing of PPAR-α signalling and decreasing fatty acids (FAs) beta-oxidation, leads to IR and related inflammation [85]. Relevantly, both the DNA methylation and histone modifications may also occur in mitochondrial DNA (mt-DNA) [86].
This feature appears in line with the aforementioned mitochondrial dysfunction implicated in MAFLD pathogenesis. Hypermethylation of mitochondrially encoded NADH ubiquinone oxidoreductase core subunit 6 (MT-ND6) was demonstrated on liver biopsy samples from NASH patients compared with subjects affected by liver steatosis [87]. In addition, the authors highlighted the association of MT-ND6 methylated/unmethylated DNA ratio with the NAFLD activity score (NAS) [87]. In keeping, a more recent study by Pirola et al. revealed higher levels of MT-Cytochrome B variance and mt-DNA damage in NASH patients compared to simple steatosis ones [86].
Regarding mt-DNA histone modifications, genetic polymorphism of SIRT3, a mitochondrial sirtuin pivotal for guaranteeing mitochondrial integrity and metabolism during oxidative stress [88], has been associated with the development of MS in mice and humans as well as contributes to downregulate autophagy, leading to lipotoxicity in hepatocytes and thus MAFLD worsening [89][90][91].
The role of epigenetics is not only limited to the DNA access variations being expressed also in post-transcriptional steps acting by miRNAs, small non-coding single strand RNAs (ssRNAs) able to repress or degrade specific mRNAs target, regulate several biological and pathological processes, and intervene in lipid metabolism and inflammatory processes [92][93]. In this context, one of the most relevant studied miRNAs is miRNA-122, identified as a promising biomarker and drug target for MAFLD [94]. Esau et al. firstly highlighted the key role of this miRNA in the liver of high-fat diet (HFD) fed mice; after treatment with antisense oligonucleotides (ASO) inhibitors of miRNA-122, a significant reduction of hepatic biosynthesis of FAs, an increased beta-oxidation, and a reduction of TG accumulation were observed [95]. Recent scientific evidence demonstrated through in vitro and animal models the miRNA-122 suppressing role of SIRT1 expression via binding its 3’-untranslated region (UTR), enhancing lipogenesis, and contributing, thus, to lipid accumulation [94].
In contrast, miRNA-122 knockdown mitigated this consequence through the upregulation of SIRT1 and the activation of the liver kinase B1 (LKB1)/adenosine monophosphate-activated protein kinase (AMPK) signalling pathway [94]. Besides miRNA-122, other miRNAs could interfere with these processes [96]: for instance, miRNA-33a/b action on the target AMPK, by reducing AMPK expression, increases the levels of intrahepatic TG [97]; contrariwise, miRNA-33a/b inhibition promotes beta-oxidation of FAs and insulin sensitivity [97][98]. However, the role of miRNAs was not only related to the induction of steatosis because of the proved effects on the progression to inflammation and fibrosis [99]. In this sense, miRNA-34a results highly expressed in MAFLD patients in a stage-dependent manner [100][101][102]. On the contrary, the expression levels of miRNA-451, able to downregulate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and tumour necrosis factor–α(TNF-α) in vitro, resulted significantly decreased in NASH patients [101].
Altogether, these epigenetic mechanisms contribute to MAFLD inter-individual variability in terms of susceptibility towards disease and its progression, influencing the clinical history of each patient. To improve MAFLD individual prognosis, the future challenge is certainly represented by the application of these findings to the routine clinical practice, through the identification of biomarkers and therapeutic targets usable for early diagnosis and personalized therapies. In these terms, the above-presented findings highlight a suggestive relationship between the occurrence of epigenetic phenomena modifying gene expression and metabolic dysfunctions (including, among others, IR and high LDL serum levels). This feature appears widely relevant, representing an important food for thought regarding potential nutrigenomics approaches considering certain nutrients’ capability to influence gene expression, discussed as well in the next paragraph.
References
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864.
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver Dis. 2018, 22, 11–21.
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415.
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pr. 2020, 35, 72–84.
- Fang, Y.-L.; Chen, H.; Wang, C.-L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model. ” World J. Gastroenterol. 2018, 24, 2974–2983.
- Taliento, A.E.; Dallio, M.; Federico, A.; Prati, D.; Valenti, L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2755.
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: A multicenter biopsy-based study. J. Lipid Res. 2017, 58, 247–255.
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment. Pharmacol. Ther. 2018, 47, 826–837.
- Dallio, M.; Sangineto, M.; Romeo, M.; Villani, R.; Romano, A.D.; Loguercio, C.; Serviddio, G.; Federico, A. Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression. Int. J. Mol. Sci. 2021, 22, 436.
- Bibbò, S.; Ianiro, G.; Dore, M.P.; Simonelli, C.; Newton, E.E.; Cammarota, G. Gut Microbiota as a Driver of Inflammation in Nonalcoholic Fatty Liver Disease. Mediat. Inflamm. 2018, 2018, 1–7.
- Nicolucci, C.; Errico, S.; Federico, A.; Dallio, M.; Loguercio, C.; Diano, N. Human exposure to Bisphenol A and liver health status: Quantification of urinary and circulating levels by LC–MS/MS. J. Pharm. Biomed. Anal. 2017, 140, 105–112.
- Lama, S.; Vanacore, D.; Diano, N.; Nicolucci, C.; Errico, S.; Dallio, M.; Federico, A.; Loguercio, C.; Stiuso, P. Ameliorative effect of Silybin on bisphenol A induced oxidative stress, cell proliferation and steroid hormones oxidation in HepG2 cell cultures. Sci. Rep. 2019, 9, 3228.
- Eslam, M.; Sarin, S.K.; Wong, V.W.-S.; Fan, J.-G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.-H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 889–919.
- Dongiovanni, P.; Valenti, L. A Nutrigenomic Approach to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2017, 18, 1534.
- De Lorenzo, A.; Noce, A.; Bigioni, M.; Calabrese, V.; Della Rocca, D.; Daniele, N.; Tozzo, C.; Renzo, L. The Effects of Italian Mediterranean Organic Diet (IMOD) on Health Status. Curr. Pharm. Des. 2010, 16, 814–824.
- Davis, C.R.; Bryan, J.; Hodgson, J.M.; Murphy, K.J. Definition of the Mediterranean Diet: A Literature Review. Nutrients 2015, 7, 9139–9153.
- Kim, K.; Hyeon, J.; Lee, S.A.; Kwon, S.O.; Lee, H.; Keum, N.; Lee, J.; Park, S.M. Role of Total, Red, Processed, and White Meat Consumption in Stroke Incidence and Mortality: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. J. Am. Hear. Assoc. 2017, 6, e005983.
- Abenavoli, L.; Boccuto, L.; Federico, A.; Dallio, M.; Loguercio, C.; Di Renzo, L.; De Lorenzo, A. Diet and Non-Alcoholic Fatty Liver Disease: The Mediterranean Way. Int. J. Environ. Res. Public Health 2019, 16, 3011.
- Dayeh, T.A.; Olsson, A.H.; Volkov, P.; Almgren, P.; Rönn, T.; Ling, C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia 2013, 56, 1036–1046.
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and Genetics in NAFLD: The Perfect Binomium. Int. J. Mol. Sci. 2020, 21, 2986.
- Ferguson, L.R.; De Caterina, R.; Görman, U.; Allayee, H.; Kohlmeier, M.; Prasad, C.; Choi, M.S.; Curi, R.; De Luis, D.A.; Gil, Á.; et al. Guide and Position of the International Society of Nutrigenetics/Nutrigenomics on Personalised Nutrition: Part 1—Fields of Precision Nutrition. Lifestyle Genom. 2016, 9, 12–27.
- Long, M.T.; Gurary, E.B.; Massaro, J.M.; Ma, J.; Hoffmann, U.; Chung, R.T.; Benjamin, E.J.; Loomba, R. Parental non-alcoholic fatty liver disease increases risk of non-alcoholic fatty liver disease in offspring. Liver Int. 2019, 39, 740–747.
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279.
- Sánchez-Torrijos, Y.; Herrojo, J.A.; Palacios, D.P.; Gallego-Durán, R.; Romero-Gómez, M.; Ampuero, J. Analysis of the burden and variability in the management of NAFLD patients in the clinical practice: Unifying the required criteria. Rev. Española Enferm. Dig. 2019, 111, 270–274.
- Rinella, M.E.; Tacke, F.; Sanyal, A.J.; Anstee, Q.M.; the Participants of the AASLD/EASL Workshop. Report on the AASLD/EASL Joint Workshop on Clinical Trial Endpoints in NAFLD. Hepatology 2019, 70, 1424–1436.
- Clarke, J.D.; Cherrington, N.J. Nonalcoholic steatohepatitis in precision medicine: Unraveling the factors that contribute to individual variability. Pharmacol. Ther. 2015, 151, 99–106.
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209.
- Choudhary, N.S.; Duseja, A. Genetic and epigenetic disease modifiers: Non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD). Transl. Gastroenterol. Hepatol. 2021, 6, 2.
- Li, Y.-Y. Genetic and epigenetic variants influencing the development of nonalcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 6546–6551.
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; A Pennacchio, L.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465.
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 574–580.
- Rae-Whitcombe, S.M.; Kennedy, D.; Voyles, M.; Thompson, M.P. Regulation of the promoter region of the human adiponutrin/PNPLA3 gene by glucose and insulin. Biochem. Biophys. Res. Commun. 2010, 402, 767–772.
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526.
- Mondul, A.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J. Nutr. 2015, 145, 1687–1691.
- Haaker, M.W.; Vaandrager, A.B.; Helms, J.B. Retinoids in health and disease: A role for hepatic stellate cells in affecting retinoid levels. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158674.
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42.
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894.
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217.
- Musso, G.; Cassader, M.; Gambino, R. PNPLA3 rs738409 and TM6SF2 rs58542926 gene variants affect renal disease and function in nonalcoholic fatty liver disease. Hepatology 2015, 62, 658–659.
- Murphy, R.C.; Folco, G. Lysophospholipid acyltransferases and leukotriene biosynthesis: Intersection of the Lands cycle and the arachidonate PI cycle. J. Lipid Res. 2019, 60, 219–226.
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.
- Luukkonen, P.K.; Zhou, Y.; Hyötyläinen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Orešič, M.; Yki-Järvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265.
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 1–10.
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356.
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; Hooft, F.V. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918.
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.G.; McPherson, S.W.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309.
- Luukkonen, P.K.; Zhou, Y.; Haridas, P.N.; Dwivedi, O.P.; Hyötyläinen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136.
- Kim, D.S.; Jackson, A.U.; Li, Y.K.; Stringham, H.M.; Kuusisto, J.; Kangas, A.J.; Soininen, P.; Ala-Korpela, M.; Burant, C.F.; Salomaa, V.; et al. Novel association of TM6SF2 rs58542926 genotype with increased serum tyrosine levels and decreased apoB-100 particles in Finns. J. Lipid Res. 2017, 58, 1471–1481.
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514.
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088.
- Dongiovanni, P.; Meroni, M.; Mancina, R.M.; Baselli, G.; Rametta, R.; Pelusi, S.; Männistö, V.; Fracanzani, A.L.; Badiali, S.; Miele, L.; et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 666–675.
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative Med. Cell. Longev. 2018, 2018, 1–14.
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454.
- Namikawa, C.; Shu-Ping, Z.; Vyselaar, J.R.; Nozaki, Y.; Nemoto, Y.; Ono, M.; Akisawa, N.; Saibara, T.; Hiroi, M.; Enzan, H.; et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatol. 2004, 40, 781–786.
- Macaluso, F.S.; Maida, M.; Petta, S. Genetic background in nonalcoholic fatty liver disease: A comprehensive review. World J. Gastroenterol. 2015, 21, 11088–11111.
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE Genotype, Parenchymal Iron Accumulation, and Liver Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2010, 138, 905–912.
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 940–950.
- Aller, R.; A De Luis, D.; Izaola, O.; Sagrado, M.G.; Conde, R.; Alvarez, T.; Pacheco, D.; Velasco, M.C. Role of -55CT polymorphism of UCP3 gene on non alcoholic fatty liver disease and insulin resistance in patients with obesity. Nutr. Hosp. 2010, 25, 572–576.
- Fares, R.; Petta, S.; Lombardi, R.; Grimaudo, S.; Dongiovanni, P.; Pipitone, R.; Rametta, R.; Fracanzani, A.L.; Mozzi, E.; Craxì, A.; et al. The UCP2 -866 G>A promoter region polymorphism is associated with nonalcoholic steatohepatitis. Liver Int. 2015, 35, 1574–1580.
- Dongiovanni, P.; Meroni, M.; Baselli, G.; Mancina, R.M.; Ruscica, M.; Longo, M.; Rametta, R.; Cespiati, A.; Pelusi, S.; Ferri, N.; et al. PCSK7 gene variation bridges atherogenic dyslipidemia with hepatic inflammation in NAFLD patients. J. Lipid Res. 2019, 60, 1144–1153.
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-TruncatingHSD17B13Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106.
- Su, W.; Mao, Z.; Liu, Y.; Zhang, X.; Zhang, W.; Gustafsson, J.-A.; Guan, Y. Role of HSD17B13 in the liver physiology and pathophysiology. Mol. Cell. Endocrinol. 2019, 489, 119–125.
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590.
- Trépo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 65, 399–412.
- European Association for the Study of The Liver; European Association for the Study of Diabetes. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402.
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Msc, A.P.C.; Carver, T.; Hartley, S.; De Villiers, C.B.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718.
- Pelusi, S.; Baselli, G.; Pietrelli, A.; Dongiovanni, P.; Donati, B.; McCain, M.V.; Meroni, M.; Fracanzani, A.L.; Romagnoli, R.; Petta, S.; et al. Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 1–10.
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782.
- Guzman, C.B.; Duvvuru, S.; Akkari, A.; Bhatnagar, P.; Battioui, C.; Foster, W.; Zhang, X.M.; Shankar, S.S.; Deeg, M.A.; Chalasani, N.; et al. Coding variants in PNPLA3 and TM6SF2 are risk factors for hepatic steatosis and elevated serum alanine aminotransferases caused by a glucagon receptor antagonist. Hepatol. Commun. 2018, 2, 561–570.
- Scorletti, E.; Bhatia, L.; McCormick, K.; Clough, G.; Nash, K.; Calder, P.; Byrne, C.D. Design and rationale of the WELCOME trial: A randomised, placebo controlled study to test the efficacy of purified long chain omega-3 fatty treatment in non-alcoholic fatty liver disease. Contemp. Clin. Trials 2014, 37, 301–311.
- Sun, C.; Fan, J.-G.; Qiao, L. Potential Epigenetic Mechanism in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2015, 16, 5161–5179.
- Hotta, K.; Kitamoto, A.; Kitamoto, T.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of differentially methylated region (DMR) networks associated with progression of nonalcoholic fatty liver disease. Sci. Rep. 2018, 8, 13567.
- Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Imajo, K.; Saito, S.; Yoneda, M.; Nakamura, T.; Nakajima, A.; Hotta, K. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 494–502.
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley–Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between Methylome and Transcriptome in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 1076–1087.
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 2015, 7, 25.
- Li, Y.Y.; Tang, D.; Du, Y.L.; Cao, C.Y.; Nie, Y.Q.; Cao, J.; Zhou, Y.J. Fatty liver mediated by peroxisome proliferator-activated receptor-α DNA methylation can be reversed by a methylation inhibitor and curcumin. J. Dig. Dis. 2018, 19, 421–430.
- Chen, H.-C.; Chen, Y.-Z.; Wang, C.-H.; Lin, F.-J. The nonalcoholic fatty liver disease-like phenotype and lowered serum VLDL are associated with decreased expression and DNA hypermethylation of hepatic ApoB in male offspring of ApoE deficient mothers fed a with Western diet. J. Nutr. Biochem. 2020, 77, 108319.
- Tyagi, S.; Sharma, S.; Gupta, P.; Saini, A.S.; Kaushal, C. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240.
- De Mello, V.D.; Matte, A.; Perfilyev, A.; Männistö, V.; Rönn, T.; Nilsson, E.; Käkelä, P.; Ling, C.; Pihlajamäki, J. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 2017, 12, 287–295.
- de Mello, V.D.; Sehgal, R.; Männistö, V.; Klåvus, A.; Nilsson, E.; Perfilyev, A.; Kaminska, D.; Miao, Z.; Pajukanta, P.; Ling, C.; et al. Serum aromatic and branched-chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int. 2021, 41, 754–763.
- Saran, A.R.; Dave, S.; Zarrinpar, A. Circadian Rhythms in the Pathogenesis and Treatment of Fatty Liver Disease. Gastroenterology 2020, 158, 1948–1966.e1.
- Mazzoccoli, G.; Vinciguerra, M.; Oben, J.; Tarquini, R.; De Cosmo, S. Non-alcoholic fatty liver disease: The role of nuclear receptors and circadian rhythmicity. Liver Int. 2014, 34, 1133–1152.
- Nassir, F.; A Ibdah, J. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092.
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338.
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; O Castaño, G.; Sookoian, S. Liver mitochondrial DNA damage and genetic variability of Cytochrome b—A key component of the respirasome—Drive the severity of fatty liver disease. J. Intern. Med. 2021, 289, 84–96.
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363.
- Kim, H.-S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 Is a Mitochondria-Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism during Stress. Cancer Cell 2010, 17, 41–52.
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome. Mol. Cell 2011, 44, 177–190.
- Yoshino, J.; Imai, S.-I. Mitochondrial SIRT3: A New Potential Therapeutic Target for Metabolic Syndrome. Mol. Cell 2011, 44, 170–171.
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003.
- Finch, M.L.; Marquardt, J.U.; Yeoh, G.C.; Callus, B.A. Regulation of microRNAs and their role in liver development, regeneration and disease. Int. J. Biochem. Cell Biol. 2014, 54, 288–303.
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207.
- Long, J.-K.; Dai, W.; Zheng, Y.-W.; Zhao, S.-P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26.
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98.
- Salvoza, N.C.; Klinzing, D.C.; Gopez-Cervantes, J.; Baclig, M.O. Association of Circulating Serum miR-34a and miR-122 with Dyslipidemia among Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0153497.
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Salinas, D.C.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237.
- Erhartova, D.; Cahova, M.; Dankova, H.; Heczkova, M.; Mikova, I.; Sticova, E.; Spicak, J.; Seda, O.; Trunecka, P. Serum miR-33a is associated with steatosis and inflammation in patients with non-alcoholic fatty liver disease after liver transplantation. PLoS ONE 2019, 14, e0224820.
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; De Sousa, M.C.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079.
- Pillai, S.S.; Lakhani, H.V.; Zehra, M.; Wang, J.; Dilip, A.; Puri, N.; O’Hanlon, K.; Sodhi, K. Predicting Nonalcoholic Fatty Liver Disease through a Panel of Plasma Biomarkers and MicroRNAs in Female West Virginia Population. Int. J. Mol. Sci. 2020, 21, 6698.
- Hur, W.; Lee, J.H.; Kim, S.W.; Kim, J.-H.; Bae, S.H.; Kim, M.; Hwang, D.; Kim, Y.S.; Park, T.; Um, S.-J.; et al. Downregulation of microRNA-451 in non-alcoholic steatohepatitis inhibits fatty acid-induced proinflammatory cytokine production through the AMPK/AKT pathway. Int. J. Biochem. Cell Biol. 2015, 64, 265–276.
- Liu, X.-L.; Pan, Q.; Zhang, R.-N.; Shen, F.; Yan, S.-Y.; Sun, C.; Xu, Z.-J.; Chen, Y.-W.; Fan, J.-G. Disease-specific miR-34a as diagnostic marker of non-alcoholic steatohepatitis in a Chinese population. World J. Gastroenterol. 2016, 22, 9844–9852.

