 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rebecca Sims | + 3678 word(s) | 3678 | 2021-05-19 05:05:33 | | | |
| 2 | Camila Xu | + 514 word(s) | 4192 | 2021-05-31 03:20:44 | | | | |
| 3 | Camila Xu | + 514 word(s) | 4192 | 2021-05-31 03:29:16 | | |
Video Upload Options
Sporadic Alzheimer’s disease (AD) is a complex genetic disease, and the leading cause of dementia worldwide. While great strides have been made in understanding the genetic aetiology of disease there remains a large proportion of the genome unexplored for complex disease.
1. Introduction
Alzheimer’s disease (AD) is the leading cause of dementia worldwide. Early family studies identified mutations in amyloid precursor protein (APP) and presenilin (PSEN) genes [1][2][3] as the cause of rare, early onset, familial forms of the disease. From this, scientists hypothesized the role of the amyloid cascade in the aetiology of sporadic AD. However, recent failures in clinical trials, based on removing either soluble and/or insoluble Aβ or targeting enzymes responsible for cleavage of APP, have thrown doubt on the hypothesis [4][5]. Familial rare mutations, that induce early onset AD (EOAD), constitute less than 1% of all AD cases. Common forms of late-onset AD (LOAD) have heritability estimates of 56–79% [6] and are contributed to by multiple genetic risk factors [7].
Over the past three decades great advances in gene discovery have been made by worldwide collaborative projects such as European AD DNA Biobank (EADB), AD Sequencing Project (ADSP), AD European Sequencing Consortium (ADES) and the International Genomics of Alzheimer’s Project (IGAP). These pioneering studies are summarized elsewhere [7]. To date around 70 genetic variants have been reported to be associated with AD risk (Figure 1) and these discoveries have indicated the involvement of pathways, additional to APP metabolism, such as immunity, ubiquitination, endocytosis, lipid metabolism and tau binding [8][9]. Furthermore, generation of large databases of genetic data have led to discoveries of genetic prediction strategies such as the polygenic risk score for AD [10].
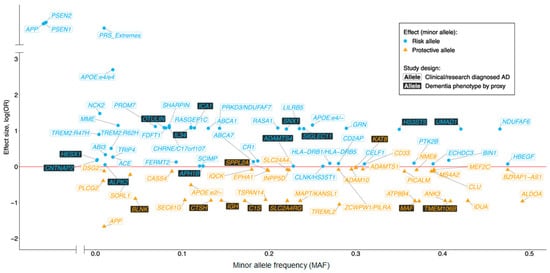
Figure 1. Schematic representation of Mendelian disease-causing genes and loci reaching genome-wide significance (GWS) for single variant (not gene-wide) association with sporadic Alzheimer’s disease (AD). Filled blue and orange points represent risk and protective association, respectively, in AD diagnosed cohorts. Adapted from Sims et al. [7] with the addition of variants identified by EADB [11].
Ultimately, the genetic research carried out to date has contributed vastly to our understanding of Alzheimer’s disease and gives hope that AD can be diagnosed and treated before the onset of irreversible neurodegeneration. However, it is estimated that a substantial proportion (approximately half) [12][13] of the genetic variance of LOAD is not yet accounted for. The majority of genetic research has focused on single nucleotide polymorphisms (SNPs), and utilized genome-wide association studies (GWAS), due the ease of genotyping in a high-throughput, cost and time effective manner. GWAS uses genotyping arrays based on known genetic variants. Therefore, the undiscovered genetic heritability is likely to be accounted for by other forms of variation, especially rare protein-coding or large variable number of tandem repeats (VNTRs) that either increase the risk of disease or offer protection against it. Next generation sequencing (NGS) has enabled researchers to screen large sample populations to detect novel rare small nucleotide variants for AD however, short read length limits the detection of larger structural variants within the DNA. Copy number variants (CNVs), inversions and translocations and VNTRs, in particular, have been unintentionally overlooked because of limited technological advances.
2. Next Generation Sequencing in AD Studies
Next Generation Sequencing (NGS) is generally based on spatially separated amplified DNA regions that cover specific sequences of genome such as the whole genome, whole exome or targeted gene regions. In short read sequencing experiments these amplified regions, known as reads, are 75 to 150 base pairs (bp) in length. The amplified regions are sequenced in a flow cell and aligned to a reference genome to detect DNA variation within the sample. The use of short sequences enables DNA sequencing at high throughput and at a high depth in order to detect mutations with high accuracy for a large sample size. The reduction in cost and increase in throughput has led to new discoveries in the field of AD genetics (Table 1). For example, in families with an autosomal dominance inheritance pattern of AD without PSEN or APP mutations, whole exome sequencing (WES) led to the discovery of novel Sortilin related receptor (SORL1) mutations. SORL1 is a protein involved in the control of amyloid production and there may be many mutations within this gene that contribute to AD with varying risk based on the damage the variant makes to the SORL1 protein and the rareness of the variant [14]. Recently, WES screening of a large AD case control sample confirmed that carrying SORL1 rare, damaging protein variants approximately triples the risk of EOAD and doubles the risk of LOAD [15]. WES has also uncovered novel damaging variants in TREM2 [16][17], IGHG3 and ZNF655 [18] and NSF [19]. The encoded proteins are functional in pathways known to confer AD risk such amyloid processing or inflammation.
Whilst NGS technology has led to discovery of variants that increase risk of AD, NGS has also identified variants that offer protection against AD in APP, CBX3 and PRSS3 [15][20]. Identification of protective variation in AD is as important as identifying disease causing variation. These protective variants highlight biological pathways that provide resilience to disease and have the potential for therapeutic intervention for improved treatment or even prevention of clinical AD.
Table 1. A summary of NGS short read sequencing studies in AD. Association statistically significant at p < 10−6 exome wide for WES and p < 10−8 genome-wide for Whole Genome Sequencing (WGS).
| Author | Method | Cohort | Cohort Size (n) | Outcome |
|---|---|---|---|---|
| [21] | Agilent SureSelect Human & Illumina sequencing | Autosomal dominant EOAD without known mutations | 14 | 7 unknown mutations detected in SORL1 gene. |
| [20] | WGS, Illumina bead chip, and imputation | Icelandic | 1795 WGS 71743 chip |
Coding mutation A673T in APP protects against AD. |
| [16] | WGS, Illumina bead chip, and imputation | Icelandic | 1795 WGS | rs75932628-T (R47H) of TREM2 increases risk of LOAD. |
| [17] | Illumina WES TruSeq, Illumina Sequenced | Caucasian from UK, US and Canada | 1092 AD cases 1107 Controls |
rs75932628-T (R47H) of TREM2 increases risk of LOAD. |
| [14] | Roche NimbleGen v2 and v3, SeqCap® EZ Exome, Illumina TruSeq | 1908 Dutch AD cases and Controls | 640 cases 1268 Controls |
181 unique SORL1 variants detected. Developed a strategy to classify SORL1 variants into five subtypes ranging from pathogenic to benign. |
| [22] | Targeted sequencing using Agilent HaloPLex™ and Illumina sequencing | Unrelated LOAD and matched controls | 772 LOAD 757 Controls |
Intronic variant rs78117248 in ABCA7 showed strongest association with AD. Loss of function mutations may be a potential pathogenic mechanism. |
| [18] | WGS and WES data generated by ADSP using Roche NimbleGen and Illumina Rapid Capture Exome | European-American and Hispanic | 172 AD 171 Controls |
Novel variants in previously reported AD risk genes and variants in novel genes IGHG3 and ZNF655 were detected. |
| [19] | Agilent SureSelect and ADSP using Roche NimbleGen and Illumina Rapid Capture Exome | Caucasian non APOE*4 carriers from PITT-ADRC 1 ADSP 2 |
PITT-ADRC1372 AD 348 Controls ADSP2 2113 AD 5139 Controls |
Association at novel variant in NSF gene and known loci in TREM2, TOMM40 and APOE was detected. |
| [15] | Roche NimbleGen, Agilent NextEra, Truseq, HaloPlex, SureSelect, Illumina Nextera® Rapid Capture Exome | 25982 Caucasian | 12652 AD 8693 controls |
Detected protein damaging variants in TREM2, SORL1 and ABCA7 and protective variants in CBX3 and PRSS3. |
| [23] | ADSP using Nimblegen NimbleGen v2 and v3 and Illumina Nextera® Rapid Capture Exome | 5142 | 4889 | Identified a VNTR within MUC6 associated with pTau burden. |
1: University of Pittsburgh Alzheimer’s Disease Research Center (ADRC). 2: Alzheimer’s Disease Sequencing Project (ADSP).
3. The Dark Genome
One of the key limitations of recent discoveries by short read NGS is that not all generated sequences are adequately mapped or aligned to the genome. This can be due to technical issues arising from poor quality of DNA or type of kit used. It is also due to the nature of the human genome. The human genome consists of many repeat elements and gene duplications and translocations making it too complex to map short read sequences with certainty (Figure 2). There are also camouflaged regions where reads cannot be mapped because the region has been duplicated in the genome. This missing element is described as the “dark genome”.
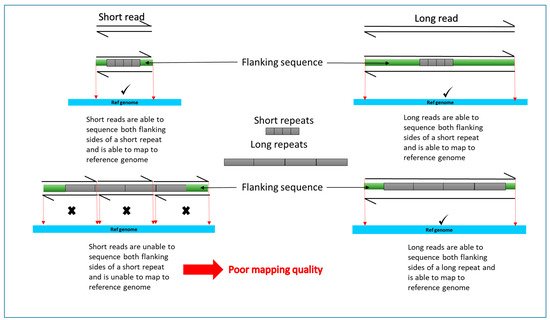
Figure 2. A simplified, schematic diagram comparing short read and long read mapping to reference genome for long and short repeats. Sequencing reads are indicated by partial arrows. Grey rectangles represent target sequence. Flanking sequence is represented by green rectangles. Reference genome used to align the reads is indicated in blue. Reads poorly or not aligned to the reference genome are indicated by X. Success in mapping to reference genome indicated by a tick.
A systematic analysis of dark regions in short read NGS data was undertaken for ten unrelated males from the ADSP [21]. The study reported that standard short-read sequencing leaves 36,794 dark regions across 6054 gene bodies, including protein coding exons from 748 genes. Sixty-two percent of the dark gene bodies are dark because of mapping quality and 71.1% of gene regions were replicated three or more times within the genome leading to poor mapping quality. When they applied long read technologies from 10x Genomics, PacBio and Oxford Nanopore Technology they reported a reduction in dark protein-coding regions to approximately 50.5%, 35.6%, and 9.6% for each technology, respectively.
4. The importance of Large Structural Variants in Complex Disease
Large structural variants arise from errors in DNA repair, recombination, translocations and result in DNA variants for example, deletions, duplications, insertions and translocations. Repeats are thought to constitute approximately 3% of the human genome [22]. Short tandem repeats (STRs) are the most commonly observed variety of repeat in the human genome. STRs such as homonucleotide, dinucleotide and trinucleotide repeats have been implicated in regulating gene expression through mechanisms such as altering splice sites, modulating binding of transcription factors and changing promoter region DNA sequences [23]. Furthermore, larger, more complex repeats for instance variable number of tandem repeats (VNTRs) and copy number variants (CNVs) are not as common as STRs but contribute to rare forms of disease with high pathogenic effect.
The 1000 Genomes project and disease sequencing studies have generated maps that allow scientists to understand how VNTRs are associated with a specific type of disease. There are many published Mendelian VNTRs and regions of genomic instability that are associated with distinct phenotypes. Deletions on 22q11 are the most common form of microdeletions in humans. The deletions on 22q11 are thought be caused by large chromosome specific low copy repeats that affect chromosomal events during meiosis [24] and result in complex phenotypes including DiGeorge Syndrome and Velo Cardio Facial syndrome (VCF). A “CGG” trinucleotide repeat within the Fragile X Mental Retardation 1 (FMR1) gene, on the X chromosome, can result in hyper-methylation and silencing of FMR1. Those who possess 55 or more copies of the repeat within the FMR1 gene tend to have a complex disorder that features intellectual disabilities, psychiatric disabilities, neuro-degenerative disorders combined with facial abnormalities. Huntington’s disease (HD) is caused by a “CAG” trinucleotide repeat within exon 1 of the Huntington (HTT) gene. HD severity increases with increasing CAG repeat length repeat length and expansions of greater than 36xCAG result in complex phenotypes including degeneration of the cortex and striatum. Furthermore, HTT CAG repeat length only accounts for up to half of the variation in age at onset [25] and disease modifiers such as FAN1 influence DNA repair and CAG expansion [26].
Uncovering repeats associated with complex disease is particularly difficult due to many genes and environmental factors synchronizing together to create a phenotype. In addition, sub-phenotypes cause heterogeneity within a disease and contribute to differing ages at onset and symptom profiles. Nucleotide repeats associated with complex phenotypes often coincide with Mendelian disease loci [27]. A study that generated a comprehensive map of 11,700 CNVs, identified 30 trait associated SNPs that were in high linkage disequilibrium with CNVs [28]. It is highly plausible that some AD genome-wide significant (GWS) hits, may be in LD with nearby CNVs or other structural variation.
Structural Variation in Dementia
Candidate gene approaches have identified a proportion of large structural DNA variation in AD (Table 2). However, these studies commonly had small sample sizes, lacked power and regularly failed to replicate. Meaning a number of associations documented in the literature warrant further investigation.
The Insulin (INS) gene possesses a tandem repeat of 15 bp which ranges from 400 bp to 8000 bp in size [29]. This VNTR was genotyped in a Caucasian population but no association with AD was reported. However, this group reported an earlier age at onset (AAO) in participants homozygous for Class III INS VNTR repeat [30] which contains 139 repeats [29].
Interleukin 6 (IL6) possesses an AT rich region, spanning over 500 bp, with 4 known alleles: A, B, C and D [31]. The D allele of the IL6 VNTR has been reported to be associated with increased risk of AD [32] and the C allele is reported to offer protection against AD and delay the onset of symptoms [32][33]. Subsequent to these publications, the role of inflammation in AD has been reported through GWAS and pathway analysis but association at IL6 has not yet been reported at the genome-wide significance level.
The Monoamine oxidase inhibitor A gene (MAOA) is located within a region of known instability on the X chromosome and plays a wide range of roles in circadian rhythm and degradation of neurotransmitters. Within the promoter region of MAOA is a 30 bp VNTR that influences the expression of MAOA. Association within the MAOA locus has not been reported by GWAS or WES studies. However, the VNTR within the promoter region of MAOA has been reported to play a in sleep disturbances in AD [34] and cognitive function in AD [35].
Nitric oxide is a potential neurotoxin and is produced by three isozymes of nitric oxide synthase (NOS). The link between AD and NOS is not clear and NOS may have both neuroprotective and neurotoxic effects [36]. NOS1 gene possesses a CA dinucleotide VNTR located in the promoter and is classified as long (more than 10 repeats) or short (10 repeats or less) in association studies. The NOS1 promoter VNTR has been reported to be associated with increased AD risk and interacts with the largest single genetic risk factor for AD, APOE ε4 allele to further increase AD risk [37].
Pathogenic repeats have also been reported to contribute to other neuro-degenerative diseases. For example, in frontal temporal dementia (FTD) and amyotrophic lateral sclerosis (ALS), a hexamer (GGCCC) repeat in C9orf72, has accounted for both familial and sporadic forms of disease [38][39][40]. The repeat, over 30 repeats in length, stretches over 180 bp and can contain insertions and deletions in the flanking regions making it difficult to sequence using short read technology and difficult to genotype using repeat primed PCR or Southern blot. The repeat expansion in C9orf72 may lead to an alternatively spliced transcript and formation of nuclear RNA foci. The repeat in C9orf72 was originally detected in ALS and FTD cohorts using Sanger sequencing [38] and recently Expansion Hunter (detailed below) was developed to enable genotyping by short length read technology [41].
Table 2. A summary of published associations of large structural variation with AD.
| Author | Gene | Polymorphism Size | N Cases | N Controls | Outcome |
|---|---|---|---|---|---|
| [36] | IL6 | 500–800 bp AT rich repeat with 4 alleles (A,B,C or D) |
102 | 191 | C allele is associated with AD protection (p = 0.025) and with delayed onset of AD (p = 0.034). |
| [33] | INS | 400–8000 bp | 58 | 161 | No association with AD (p = 0.873). Class III INS VNTR homozygotes had earlier AAO (p = 0.0002). |
| [35] | IL6 | 500–800 bp AT rich repeat with 4 alleles (A,B,C or D) | 184 | 273 | C allele was negatively associated with AD (p = 0.001), DD genotype associated with AD (p = 0.015; OR = 1.636, 95%CI = 1.101-2.432). Patients with D allele of VNTR had higher plasma levels of IL6 (p = 0.001). |
| [37] | MAOA | 30 bp repeat. Alleles with 3, 3.5, 4 and 5 repeats | 425 | n/a | 4 repeats associated with sleep disturbance in AD patients (p = 0.005) |
| [42] | MAOA | 30 bp repeat. Alleles with 3, 3.5, 4 and 5 repeats | 44 | 63 | 3.5 and 4 repeats associated with increased expression of MAOA in AD pineal glands. |
| [38] | MAOA | 30 bp repeat. Alleles with 3, 3.5, 4 and 5 repeats | 193 | n/a | Sex- genotype interaction may determine cognitive scores |
| [40] | NOS1 | CA repeat Long >10 repeats Short 10 repeats |
184 | 144 | The short allele of NOS1 VNTR is associated with AD (p = 0.0009) and interact with APOEε4 allele to increase risk. |
| [43] | ABCA7 | 25 bp repeat 12–247 repeats |
An increase in VNTR length was associated with decreased ABCA7 expression. Increased VNTR length was associated with alternative splicing. | ||
| [23] | MUC6 | VNTR exon 31 Length 9–12 kbp |
119 Cohort 173 Replication |
n/a | pTau pathology associated with increased VNTR length (p = 0.031) |
5. Shedding Light on the Dark Genome
As mentioned above, short read technologies are unable to detect large structural variants due to poor mapping quality (Figure 2). Current technology and bioinformatics are changing to enable us to fill in the heritability gaps created by the dark genome. More detailed analysis of short read data could potentially lead to detection of AD risk or protective variants hidden within dark regions of data.
5.1. Generic Molecular Biology Approaches
Where NGS has identified regions that poorly map to the genome, some studies have applied “traditional” molecular biology methods such as Sanger sequencing, long range PCR and Southern blot to further characterize genomic locations indicative of association with AD risk:
Katsumata et al. [44] used SKAT-O to analyse short read WES data, generated by ADSP, to identify novel rare risk variants for LOAD. SKAT-O detected both positively and negatively associated variants within the Mucin6 gene (MUC6) however, association at these variants did not pass quality control due to poor sequence mapping to the human genome. The variants within MUC6 were within a region with complex tandem repeats and further inspection, using long range PCR, cloning, Sanger sequencing and restriction digestion identified a highly polymorphic VNTR within MUC6. This group then explored the relationship between AD neuropathology in autopsied individuals and reported that individuals with longer VNTR regions had significantly more pTau burden.
There is a similar story for the ATP-Binding Cassette Subfamily A Member 7 gene (ABCA7). Premature termination codons (PTCs) are 4 to 5 times more prevalent in ABCA7 in AD patients however, until recently, the functional variant that accounted for the GWAS signal remained undetected. Targeted gene sequencing, using short read technology, detected association at PTCs [45] but these still did not explain the GWAS association. Sanger sequencing of a stretch of DNA up to 3 kbp identified a 25 bp repeat within the splice donor site of exon 18 in ABCA7. Subsequent genotyping using Southern blot showed that individuals carrying an allele > 5720 bp (“expanded”), were mostly AD patients [OR 4.5 (95% CI 1.3–24.2), p = 0.008 [46].
Methods such as long-range PCR, Sanger sequencing and Southern blot are successful in characterizing and genotyping long stretches of DNA harbouring complex repeat elements. Unfortunately, these methods are not high throughput or time efficient and require large quantities of un-degraded DNA.
5.2. Bioinformatics Approaches
A number of studies have attempted to investigate VNTRs by applying novel bioinformatics approaches to short read NGS data. Ebbert et al. [21] developed an algorithm to extract all regions within gene bodies with a mapping quality (MAPQ) score less than 10 and then identified the regions using BLAT [42]. This algorithm was applied to the ADSP data in an attempt elucidate most of these dark regions. They identified a frameshift deletion in C3b and C4b binding region of known AD risk gene Complement Receptor 1 (CR1) but further work is required to establish this association. Ebbert et al. [21] also identified dark regions within additional gene bodies, linked with AD association including ABCA7, INPP5D, IQCK and HLA.
Recursive exact matching is a method that can detect large regions of repeats within sequence data [43]. The REVEAL algorithm utilizes recursive exact matching and creates a hierarchal tree of decisions based on maximally unique matching (MUM). The REVEAL algorithm gives a high resolution, enables alignments of repetitive sequence data and can detect inversions and translocations [43].
Imputation tools have been developed that can combine datasets taken from NGS and SNP genotyping panels to predict the genotype of short tandem repeats (STRs). Saini et al. [47] generated a catalogue of STR genotypes from a cohort of families using NGS (150 bp read length). They then imputed the STR genotypes into 1000 Genomes SNP data [48] to create a SNP+STR haplotype reference panel. The reference panel generated can be used to impute STRs into larger genetic datasets such as AD cohorts where SNP genotype data exist for tens of thousands of samples. The haplotype panel created by Saini et al. (2018) is limited in that it can only accurately predict STRs that are fully spanned by a read. It cannot detect or predict long and more complex repeats due to the read length not spanning the full length of the repeat.
Tang et al. [49] developed TREDPARSE, to identify each allele length at predefined STR loci by using Illumina WGS sequence data (read length 150 bp) that are sampled at sufficient depth. TREDPARSE is based on 4 models of calling STRs: model 1 is based on a read that spans the entire repeat and flanking region; model 2 is based on partial reads of the repeat including flanking regions; model 3 is based on reads that consist of repeat only; model 4 is based on paired end reads where one read flanks the beginning of a repeat and the paired read flanks the end of a repeat. These 4 modes allow the allele calling of both long and short repeats on short read NGS data.
Expansion Hunter is a tool that can be used to detect both long and short repeats [41]. Expansion Hunter catalogues repeats associated with the repeat of interest and approximates the size of the repeat based on the flanking reads. A maximum likelihood genotype is created by the spanning, flanking and in-repeat reads. Expansion Hunter works well for repeats because it relies on the basis that all repeats are identical. Expansion Hunter does not work as well for VNTRs due to the complex variation between repeat units.
adVNTR [50] was developed for genotyping VNTRs at targeted loci in a donor genome. For any target VNTR in a donor, adVNTR reports an estimate of repeat unit counts and point mutations within the repeat units. Hidden Markov Models are trained using reference assemblies as well as training of the flanking regions of VNTRs in the reference genome.
The above approaches can be applied to currently existing AD genotype and sequencing datasets to explore the genome for additional structural variants that contribute to the heritability of AD. However, even with these improvements in bioinformatics methodology there still remain large structural variants that are too large to detect using short read NGS technology.
5.3. Long Read Sequencing Technologies
Long read technologies offer a means to examine the genome with high resolution with read lengths up to 80 kbp or larger. Long read sequencing is slowly becoming more affordable and higher throughput. Therefore, long read sequencing is becoming a more favourable method for detecting and genotyping large structural variants. There are three main providers for long read sequencing supplying to the market. These are assessed in detail elsewhere [51] and reviewed in brief here.
The SMRT Sequel II is a recent release from PacBio. The Sequel II can sequence DNA strands over 15 kilobases in length. SMRT Sequel II utilizes SMRTbell technology, which is created by ligating hairpin adaptors to both ends of a target double stranded DNA molecule [52]. A polymerase is used to replicate the full length of the DNA strand allowing many copies of the full-length strand to be replicated. Multiple sequenced copies of each strand allow higher accuracy of nucleotide calling and reduces errors in reads. It is possible to multiplex samples using adapter primers reducing cost of sequencing. This technology is ideal for detecting detect structural variants (SVs), CNVs, and large indels ranging in size from tens to thousands of base pairs.
Oxford Nanopore Technologies (ONT) offer a number of platforms for long read sequencing: The MinION, GridION and PromethION can produce extremely long reads up to 4 Mbp in length. The ONT platforms perform sequencing of a DNA strand based on ionic changes in current within a flow cell. DNA, with DNA-protein adapters tightly bound to a polymerase, is placed in a flow cell with ionic solutions. The polymerase allows stepwise unwinding and movement of the DNA strand through a pore and ionic current changes are measured as the DNA strand passes through. The ionic current changes are used to construct a “squiggle plot” that is processed by minKNOW software into DNA sequence data. The MinION is a small, portable device and is suitable for work in the field. The GridION and PromethION are designed for larger scale, high-throughput projects. Whilst ONT sequencers can sequence very long strands of DNA, the DNA reads are based on up to two copies of the strand. Therefore, this method of sequencing can have a high error rate of base calling.
An alternative approach to long read whole genome sequencing is offered by 10X Genomics. The Chromium™ Genome Protocol generates long-range information across the length of individual DNA molecules with a mean length greater than 65 kbp. Using a microfluidics chip, single strands of template genomic DNA are encapsulated in separate Gel Bead-In-Emulsions (GEMs) containing copies of a unique barcoded primer. Once the DNA strands are partitioned within a GEM and labelled with unique barcodes specific for that GEM, the fragments are sheared and sequenced using classical Illumina short read technology. The weakness of this approach is that it utilizes PCR and increases the risk of PCR based errors. Therefore, deep sequencing of greater than 30x copies is required.
Ultimately, what determines the success of long read sequencing experiments, is DNA quality. Intact, un-degraded and pure DNA is required for these methods. Ideally, fresh DNA that has not been subjected to freeze-thaw or fixation is a prerequisite. Currently, large DNA banks possess tens of thousands of DNA samples collected over the past 15 years or more and many of these samples would be deemed unsuitable for long read sequencing due to DNA degradation or impurity.
References
- Pottier, C.; Hannequin, D.; Coutant, S.; Rovelet-Lecrux, A.; Wallon, D.; Rousseau, S.; Legallic, S.; Paquet, C.; Bombois, S.; Pariente, J.; et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry 2012, 17, 875–879.
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer′s disease and age-related cognitive decline. Nature 2012, 488, 96–99.
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer′s Disease. N. Engl. J. Med. 2013, 368, 107–116.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer′s disease. N. Engl. J. Med. 2013, 368, 117–127.
- Holstege, H.; van der Lee, S.J.; Hulsman, M.; Wong, T.H.; van Rooij, J.G.J.; Weiss, M.; Louwersheimer, E.; Wolters, F.J.; Amin, N.; Uitterlinden, A.G.; et al. Characterization of pathogenic SORL1 genetic variants for association with Alzheimer′s disease: A clinical interpretation strategy. Eur. J. Hum. Genet. 2017, 25, 973–981.
- Cuyvers, E.; De Roeck, A.; Van den Bossche, T.; Van Cauwenberghe, C.; Bettens, K.; Vermeulen, S.; Mattheijssens, M.; Peeters, K.; Engelborghs, S.; Vandenbulcke, M.; et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer′s disease patients: A targeted resequencing study. Lancet Neurol. 2015, 14, 814–822.
- Bis, J.C.; Jian, X.Q.; Kunkle, B.W.; Chen, Y.N.; Hamilton-Nelson, K.L.; Bush, W.S.; Salerno, W.J.; Lancour, D.; Ma, Y.Y.; Renton, A.E.; et al. Whole exome sequencing study identifies novel rare and common Alzheimer′s-Associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 2020, 25, 1859–1875.
- Fan, K.H.; Feingold, E.; Rosenthal, S.L.; Demirci, F.Y.; Ganguli, M.; Lopez, O.L.; Kamboh, M.I. Whole-Exome Sequencing Analysis of Alzheimer′s Disease in Non-APOE*4 Carriers. J. Alzheimers Dis. 2020, 76, 1553–1565.
- Holstege, H.; Hulsman, M.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Grozeva, D.; van Rooij, J.; Sims, R.; Ahmad, S.; Amin, N.; et al. Exome sequencing identifies novel AD-associated genes. MedRxiv 2021.
- Katsumata, Y.; Fardo, D.W.; Bachstetter, A.D.; Artiushin, S.C.; Wang, W.X.; Wei, A.; Brzezinski, L.J.; Nelson, B.G.; Huang, Q.W.; Abner, E.L.; et al. Alzheimer Disease Pathology-Associated Polymorphism in a Complex Variable Number of Tandem Repeat Region Within the MUC6 Gene, Near the AP2A2 Gene. J. Neuropathol. Exp. Neurol. 2020, 79, 3–21.
- Papassotiropoulos, A.; Bagli, M.; Jessen, F.; Bayer, T.A.; Maier, W.; Rao, M.L.; Heun, R. A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk for sporadic Alzheimer′s disease. Ann. Neurol. 1999, 45, 666–668.
- Majores, M.; Kolsch, H.; Bagli, M.; Ptok, U.; Kockler, M.; Becker, K.; Rao, M.L.; Maier, W.; Heun, R. The insulin gene VNTR polymorphism in Alzheimer′s disease: Results of a pilot study. J. Neural Transm. 2002, 109, 1029–1034.
- Licastro, F.; Grimaldi, L.M.E.; Bonafe, M.; Martina, C.; Olivieri, F.; Cavallone, L.; Giovanietti, S.; Masliah, E.; Franceschi, C. Interleukin-6 gene alleles affect the risk of Alzheimer′s disease and levels of the cytokine in blood and brain. Neurobiol. Aging 2003, 24, 921–926.
- Craig, D.; Hart, D.J.; Passmore, A.P. Genetically increased risk of sleep disruption in Alzheimer′s disease. Sleep 2006, 29, 1003–1007.
- Wu, Y.H.; Fischer, D.F.; Swaab, D.F. A promoter polymorphism in the monoamine oxidase A gene is associated with the pineal MAOA activity in Alzheimer′s disease patients. Brain Res. 2007, 1167, 13–19.
- Chang, H.I.; Chang, Y.T.; Tsai, S.J.; Huang, C.W.; Hsu, S.W.; Liu, M.E.; Chang, W.N.; Lien, C.Y.; Huang, S.H.; Lee, C.C.; et al. MAOA-VNTR Genotype Effects on Ventral Striatum-Hippocampus Network in Alzheimer′s Disease: Analysis Using Structural Covariance Network and Correlation with Neurobehavior Performance. Mol. Neurobiol. 2019, 56, 4518–4529.
- Galimberti, D.; Scarpini, E.; Venturelli, E.; Strobel, A.; Herterich, S.; Fenoglio, C.; Guidi, I.; Scalabrini, D.; Cortini, F.; Bresolin, N.; et al. Association of a NOS1 promoter repeat with Alzheimer′s disease. Neurobiol. Aging 2008, 29, 1359–1365.
- De Roeck, A.; Duchateau, L.; Van Dongen, J.; Cacace, R.; Bjerke, M.; Van den Bossche, T.; Cras, P.; Vandenberghe, R.; De Deyn, P.P.; Engelborghs, S.; et al. An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer′s disease. Acta Neuropathol. 2018, 135, 827–837.
- Fan, K.H.; Feingold, E.; Rosenthal, S.L.; Demirci, F.Y.; Ganguli, M.; Lopez, O.L.; Kamboh, M.I. Whole-Exome Sequencing Analysis of Alzheimer′s Disease in Non-APOE*4 Carriers. J. Alzheimers Dis. 2020, 76, 1553–1565.
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer′s disease and age-related cognitive decline. Nature 2012, 488, 96–99.
- Ebbert, M.T.W.; Jensen, T.D.; Jansen-West, K.; Sens, J.P.; Reddy, J.S.; Ridge, P.G.; Kauwe, J.S.K.; Belzil, V.; Pregent, L.; Carrasquillo, M.M.; et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 2019, 20, 97.
- Hannan, A.J. Tandem Repeats and Repeatomes: Delving Deeper into the ‘Dark Matter’ of Genomes. EBioMedicine 2018, 31, 3–4.
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.Y.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2016, 48, 22–29.
- Saitta, S.C.; Harris, S.E.; Gaeth, A.P.; Driscoll, D.A.; McDonald-McGinn, D.M.; Maisenbacher, M.K.; Yersak, J.M.; Chakraborty, P.K.; Hacker, A.M.; Zackai, E.H.; et al. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum. Mol. Genet. 2004, 13, 417–428.
- Gusella, J.F.; MacDonald, M.E.; Lee, J.M. Genetic modifiers of Huntington′s disease. Mov. Disord. 2014, 29, 1359–1365.
- Goold, R.; Flower, M.; Moss, D.H.; Medway, C.; Wood-Kaczmar, A.; Andre, R.; Farshim, P.; Bates, G.P.; Holmans, P.; Jones, L.; et al. FAN1 modifies Huntington′s disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet. 2019, 28, 650–661.
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet. 2013, 14, 125–138.
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712.
- Rotwein, P.; Yokoyama, S.; Didier, D.K.; Chirgwin, J.M. Genetic analysis of the hypervariable region flanking the human insulin gene. Am. J. Hum. Genet. 1986, 39, 291–299.
- Majores, M.; Kolsch, H.; Bagli, M.; Ptok, U.; Kockler, M.; Becker, K.; Rao, M.L.; Maier, W.; Heun, R. The insulin gene VNTR polymorphism in Alzheimer′s disease: Results of a pilot study. J. Neural Transm. 2002, 109, 1029–1034.
- Bowcock, A.M.; Ray, A.; Erlich, H.; Sehgal, P.B. Rapid detection and sequencing of alleles in the 3′ flanking region of the interleukin-6 gene. Nucleic Acids Res. 1989, 17, 6855–6864.
- Licastro, F.; Grimaldi, L.M.E.; Bonafe, M.; Martina, C.; Olivieri, F.; Cavallone, L.; Giovanietti, S.; Masliah, E.; Franceschi, C. Interleukin-6 gene alleles affect the risk of Alzheimer′s disease and levels of the cytokine in blood and brain. Neurobiol. Aging 2003, 24, 921–926.
- Papassotiropoulos, A.; Bagli, M.; Jessen, F.; Bayer, T.A.; Maier, W.; Rao, M.L.; Heun, R. A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk for sporadic Alzheimer′s disease. Ann. Neurol. 1999, 45, 666–668.
- Craig, D.; Hart, D.J.; Passmore, A.P. Genetically increased risk of sleep disruption in Alzheimer′s disease. Sleep 2006, 29, 1003–1007.
- Chang, H.I.; Chang, Y.T.; Tsai, S.J.; Huang, C.W.; Hsu, S.W.; Liu, M.E.; Chang, W.N.; Lien, C.Y.; Huang, S.H.; Lee, C.C.; et al. MAOA-VNTR Genotype Effects on Ventral Striatum-Hippocampus Network in Alzheimer′s Disease: Analysis Using Structural Covariance Network and Correlation with Neurobehavior Performance. Mol. Neurobiol. 2019, 56, 4518–4529.
- Dubey, H.; Gulati, K.; Ray, A. Alzheimer′s Disease: A Contextual Link with Nitric Oxide Synthase. Curr. Mol. Med. 2020, 20, 505–515.
- Galimberti, D.; Scarpini, E.; Venturelli, E.; Strobel, A.; Herterich, S.; Fenoglio, C.; Guidi, I.; Scalabrini, D.; Cortini, F.; Bresolin, N.; et al. Association of a NOS1 promoter repeat with Alzheimer′s disease. Neurobiol. Aging 2008, 29, 1359–1365.
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256.
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268.
- Gijselinck, I. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65.
- Dolzhenko, E.; van Vugt, J.; Shaw, R.J.; Bekritsky, M.A.; van Blitterswijk, M.; Narzisi, G.; Ajay, S.S.; Rajan, V.; Lajoie, B.R.; Johnson, N.H.; et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017, 27, 1895–1903.
- Katsumata, Y.; Fardo, D.W.; Bachstetter, A.D.; Artiushin, S.C.; Wang, W.X.; Wei, A.; Brzezinski, L.J.; Nelson, B.G.; Huang, Q.W.; Abner, E.L.; et al. Alzheimer Disease Pathology-Associated Polymorphism in a Complex Variable Number of Tandem Repeat Region Within the MUC6 Gene, Near the AP2A2 Gene. J. Neuropathol. Exp. Neurol. 2020, 79, 3–21.
- Cuyvers, E.; De Roeck, A.; Van den Bossche, T.; Van Cauwenberghe, C.; Bettens, K.; Vermeulen, S.; Mattheijssens, M.; Peeters, K.; Engelborghs, S.; Vandenbulcke, M.; et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer′s disease patients: A targeted resequencing study. Lancet Neurol. 2015, 14, 814–822.
- De Roeck, A.; Duchateau, L.; Van Dongen, J.; Cacace, R.; Bjerke, M.; Van den Bossche, T.; Cras, P.; Vandenberghe, R.; De Deyn, P.P.; Engelborghs, S.; et al. An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer′s disease. Acta Neuropathol. 2018, 135, 827–837.
- Kent, W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664.
- Linthorst, J.; Holstege, H.; Reinders, M.J.T. Scalable multi whole-genome alignment using recursive exact matching. BioRxiv 2015.
- Saini, S.; Mitra, I.; Mousavi, N.; Fotsing, S.F.; Gymrek, M. A reference haplotype panel for genome-wide imputation of short tandem repeats. Nat. Commun. 2018, 9, 4397.
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74.
- Tang, H.B.; Kirkness, E.F.; Lippert, C.; Biggs, W.H.; Fabani, M.; Guzman, E.; Ramakrishnan, S.; Lavrenko, V.; Kakaradov, B.; Hou, C.; et al. Profiling of Short-Tandem-Repeat Disease Alleles in 12,632 Human Whole Genomes. Am. J. Hum. Genet. 2017, 101, 700–715.
- Bakhtiari, M.; Shleizer-Burko, S.; Gymrek, M.; Bansal, V.; Bafna, V. Targeted genotyping of variable number tandem repeats with adVNTR. Genome Res. 2018, 28, 1709–1719.
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681.
- Voskoboynik, A.; Neff, N.F.; Sahoo, D.; Newman, A.M.; Pushkarev, D.; Koh, W.; Passarelli, B.; Fan, H.C.; Mantalas, G.L.; Palmeri, K.J.; et al. The genome sequence of the colonial chordate, Botryllus schlosseri. Elife 2013, 2, e00569.


