Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andreja Figurek | + 1424 word(s) | 1424 | 2021-05-25 10:59:06 | | | |
| 2 | Vicky Zhou | Meta information modification | 1424 | 2021-05-31 06:24:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Figurek, A. Fibroblast Growth Factor-23. Encyclopedia. Available online: https://encyclopedia.pub/entry/10216 (accessed on 09 August 2026).
Figurek A. Fibroblast Growth Factor-23. Encyclopedia. Available at: https://encyclopedia.pub/entry/10216. Accessed August 09, 2026.
Figurek, Andreja. "Fibroblast Growth Factor-23" Encyclopedia, https://encyclopedia.pub/entry/10216 (accessed August 09, 2026).
Figurek, A. (2021, May 28). Fibroblast Growth Factor-23. In Encyclopedia. https://encyclopedia.pub/entry/10216
Figurek, Andreja. "Fibroblast Growth Factor-23." Encyclopedia. Web. 28 May, 2021.
Copy Citation
Fibroblast growth factor-23 (FGF23) appears to be one of the most promising biomarkers and predictors of cardiovascular risk in patients with heart disease and normal kidney function, but moreover in those with chronic kidney disease (CKD).
FGF23
FGF signaling
klotho
Wnt pathway
cardiomyocyte
1. FGF23 Signaling in the Physiological Milieu
FGF23 is a phosphaturic hormone primarily secreted by osteocytes to maintain phosphate and mineral homeostasis. It stimulates phosphate excretion, inhibits PTH secretion, and decreases active vitamin D levels [1][2]. FGF23 was portrayed as a crucial player in the pathogenesis of a variety of hypophosphatemic disorders, including autosomal dominant hypophosphatemic rickets, tumor-induced osteomalacia, and X-linked hypophosphatemic rickets [3].
Osteocytes and osteoblasts mainly produce FGF23. However, low FGF23 messenger RNA (mRNA) levels are detected in the brain, thymus, spleen, small intestine, heart, and testis [4].
It belongs to the superfamily of fibroblast growth factors (FGF) that exert pleiotropic effects on a vast range of biological processes, including the development of organogenesis and metabolism, being an essential part of the endocrine subfamily. While not all FGFs express endogenously in the heart of humans and rodents, FGF2, FGF3, FGF8, FGF9, FGF10, FGF16, FGF15/19, FGF21, and FGF23 act on the heart in a paracrine or endocrine manner inducing physiological or pathological pathways in the heart development, health, and disease [5]. While the paracrine FGFs are not secreted and do not exert their biological functions within the same cell, endocrine FGFs act as endocrine hormones in targeted tissues [6].
The endocrine family consists of a unique structure lacking conserved heparin (HS)-binding domain, which favors their release from the production source, disadvantaging their FGF receptors (FGFR) activation at targeted organs. They overcome this handicap, binding FGFR by the co-receptor klotho. It seems that the Klotho family of membrane proteins may have somehow evolved to increase the affinity of endocrine FGFs to FGFRs at their target organs [7].
Klotho is a membrane protein that divides into three subtypes: α-Klotho, β-Klotho, and γ-Klotho. Although the α- and β-Klotho interact with FGF23 and FGF19/FGF21, respectively, the function of the third member, γ-Klotho, continues to be unclear. FGF23 activates FGFR 1c, –3c, and –4, but not FGFR2, in the presence of cell surface membrane α-Klotho [8].
FGF23 gene encodes 32-kDa glycoprotein of 251 amino acids. The measurement of the plasma half-life of the biologically active form of FGF is reported to be 45–60 min in humans [9]. The biologically active form consists of two parts: N-terminus FGF core homology domain (155 amino acids) that shares homologies with the other FGF family members and a C-terminal domain (72 amino acids), which is essential for interaction with the FGFR-Klotho complex [9][10].
2. FGF23 Signaling in the Uremic Milieu
2.1. Cardiac Hypertrophy
As patients with CKD are at higher CV risk compared to the healthy population, it is important to understand underlying pathophysiological mechanisms and, subsequently, reduce the mentioned risk. Thus, a reliable biomarker that could estimate CV risk and mortality should be established. Hence, FGF23 was found as a sensitive biomarker elevated in early CKD [11] suitable for timely estimation of CV risk and therapeutical response in order to possibly prevent CV events and decrease mortality in those patients. Nowadays, many clinical trials reported that FGF23 is very close to being introduced into the clinical routine.
Although primarily secreted by osteocytes in bones, CKD patients have higher circulated FGF23 levels mainly due to the declining glomerular filtration and excretion by the kidneys [12][13]. In addition, increased FGF23 production in CKD (due to the impact of secondary hyperparathyroidism, treatment with active vitamin D metabolites, etc.) may contribute to the increased FGF23 plasma levels.
The pathological process starts with binding the FGF23 to its receptor in cardiomyocyte, leading to cardiac remodeling and hypertrophy that could be clinically detected. Therefore and not surprisingly, the prevalence of LVH in the general population is about 15–20% [14], whereas in the CKD population, it’s more than 70% [15].
Although experimental data in mice indicated the relative expression of FGFR3 and FGFR1 in isolated adult ventricular cardiomyocytes is higher compared to FGFR4 [16], the major functional significance in cardiac hypertrophy development seems to belong to FGFR4. Even though previously αKlotho was thought not to be expressed in cardiomyocytes, there are some experimental data indicating still very low expression [16], detected through a very high threshold cycle for αKlotho mRNA levels in the PCR analysis and with no confirmation with in situ hybridization and immunohistochemistry. This would support the fact that the role of αKlotho is not crucial in FGF23 signaling in cardiomyocytes.
It is important to underline that high FGF23 levels induce upregulation of FGFR4 expression, indicating the induction of cardiac hypertrophy via FGFR4 [17]. On the other hand, experimental studies showed that, even on a high phosphate diet, cardiac hypertrophy does not occur when FGFR4 is blocked [18]. Moreover, in experimental settings, cardiac hypertrophy is shown to be reversible [18]. Additionally, even Klotho deficiency, which also exists in CKD, contributes to cardiac hypertrophy development [19].
A generally accepted understanding is that FGF23 binds to the FGFR4 and exerts its mechanism, which is Klotho independent (Figure 1). This binding results in FGFR4 auto-phosphorylation and subsequent binding of phospholipase C y (PLCy) to the phosphorylated FGFR4 sequence activating PLCy [20]. Activated PLCy results in calcineurin activation and nuclear factor of activated T-cells (NFAT) dephosphorylation, which is then translocated to the nucleus with ensuing transcription of pro-hypertrophic genes (brain natriuretic peptide (BNP), regulator of calcineurin 1 (Rcan1), β-myosin heavy chain (β-MHC), atrial natriuretic peptide (ANP), etc.) [21][22][23][24][25]. Of note, ANP and BNP are well-defined markers of cardiac hypertrophy [26].
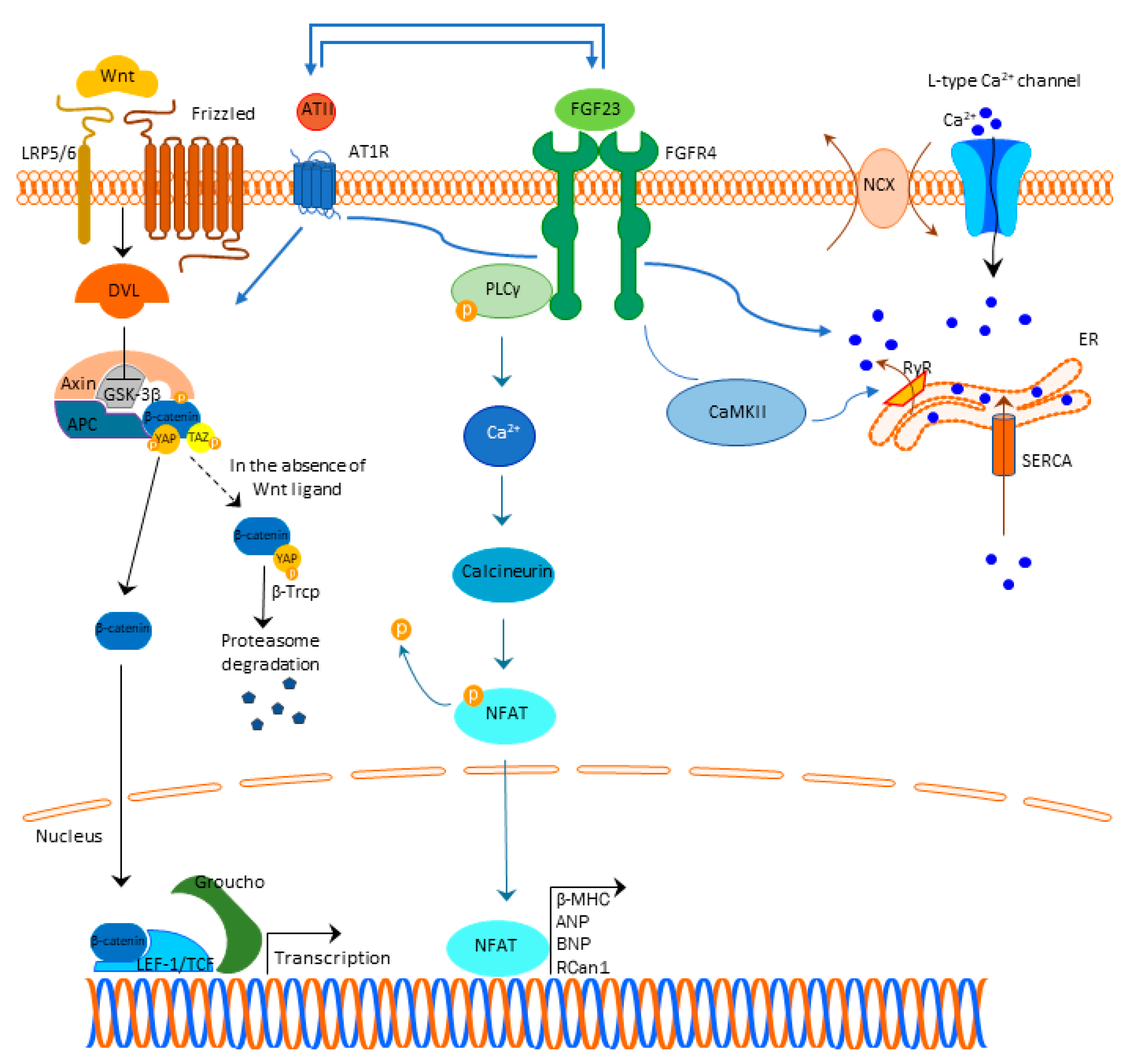 Figure 1. FGF23 signaling and cardiac hypertrophy. Abbreviations: FGF23-fibroblast growth factor 23, FGFR4—fibroblast growth factor receptor 4, PLCy—phospholipase C y, Ca2+—calcium, NFAT—nuclear factor of activated T-cells, β-MHC—β-myosin heavy chain (β-MHC), ANP—atrial natriuretic peptide, BNP—brain natriuretic peptide, Rcan1—regulator of calcineurin 1, LRP 5/6—lipoprotein-receptor-related proteins 5 and 6, DLV—Disheveled protein, APC—Adenomatous Polyposis Coli, GSK3β—glycogen synthase kinase 3β, YAP—Yes-associated protein, TAZ—transcriptional coactivator with PDZ-binding domain, β-Trcp—beta-transducin repeat containing E3 ubiquitin protein ligase, LEF/TCF—lymphoid enhancer-binding factor/β-catenin-T-cell factor, ATII—angiotensin II, AT1R—angiotensin II receptor type 1, NCX—sodium-calcium exchanger, ER—endoplasmic reticulum, RyR—ryan
Figure 1. FGF23 signaling and cardiac hypertrophy. Abbreviations: FGF23-fibroblast growth factor 23, FGFR4—fibroblast growth factor receptor 4, PLCy—phospholipase C y, Ca2+—calcium, NFAT—nuclear factor of activated T-cells, β-MHC—β-myosin heavy chain (β-MHC), ANP—atrial natriuretic peptide, BNP—brain natriuretic peptide, Rcan1—regulator of calcineurin 1, LRP 5/6—lipoprotein-receptor-related proteins 5 and 6, DLV—Disheveled protein, APC—Adenomatous Polyposis Coli, GSK3β—glycogen synthase kinase 3β, YAP—Yes-associated protein, TAZ—transcriptional coactivator with PDZ-binding domain, β-Trcp—beta-transducin repeat containing E3 ubiquitin protein ligase, LEF/TCF—lymphoid enhancer-binding factor/β-catenin-T-cell factor, ATII—angiotensin II, AT1R—angiotensin II receptor type 1, NCX—sodium-calcium exchanger, ER—endoplasmic reticulum, RyR—ryan2.2. Cardiac Fibrosis
Fibrosis is a process characterized by the production and deposition of an extracellular matrix, that can occur in any tissue or organ. Although it might be protective to a certain extent (for instance, after myocardial infarction when replacing the dead cells or during a scar-formation), it expands in the tissues increasing the mechanical pressure on it and devastating normal functional tissue.
Besides the role in cardiac hypertrophy, current findings indicate that FGF23 is involved in the pathophysiological signaling pathways that result in the development of cardiac fibrosis. Experimental studies in rats pointed out that left ventricular fibrosis did not correlate with FGFR4 and NFAT activation, indicating that the fibrotic process was not mediated through the activation of the FGF23/FGFR4/calcineurin/NFAT pathway [27]. This finding underlines the significance of the other signaling pathways, that are involved in the cardiac fibrosis occurrence, primarily FGF23/Klotho, TGFβ-, Wnt-signaling, and local RAAS.
Unlike cardiac hypertrophy, where the main pathological place occurs in cardiomyocytes, in cardiac fibrosis in addition to cardiomyocytes, the role of fibroblasts appears as crucial. In addition, although FGF23 exerts its mechanism of action by binding to the FGFR4 in cardiomyocytes, in cardiac fibroblasts, FGF23 additionally regulates TGFβ1-signaling by acting on its fibroblast growth factor receptor-1 (FGFR1) expressed in fibroblasts [28]. Experimental studies indicated that FGF23 promotes the proliferation of cardiac fibroblasts and myocardial fibrosis, both associated with the upregulation of active β-catenin and TGF-β [29].
Cardiac damage may lead to a local synthesis of FGF23 in the heart with fibroblasts as main producers increasing their proliferation, migration, and the expression of β-catenin, TGFβ1, fibronectin, and collagen I [29][30].
The cross-talk between TGFβ- and Wnt-signaling is bidirectional. TGFβ1-signaling can induce the expression of β-catenin superfamily members and β-catenin makes a complex with Smads in the nucleus, which stimulates the transcription of profibrotic genes [31]. TGFβ is able to deactivate glycogen synthase kinase 3β (GSK3β) directly and to activate Wnt/β-catenin signaling through the production of Wnt proteins [32]. Activated Wnt/β-catenin also stabilizes TGFβ/Smad response, making this co-activation of the two pathways effective in triggering the fibrotic response (Figure 2) [32]. Detailed cross-talk between TGF-β, Wnt, and YAP/TAZ is beyond the scope of this review but is nicely summarized in the review of Piersma et al. [33].
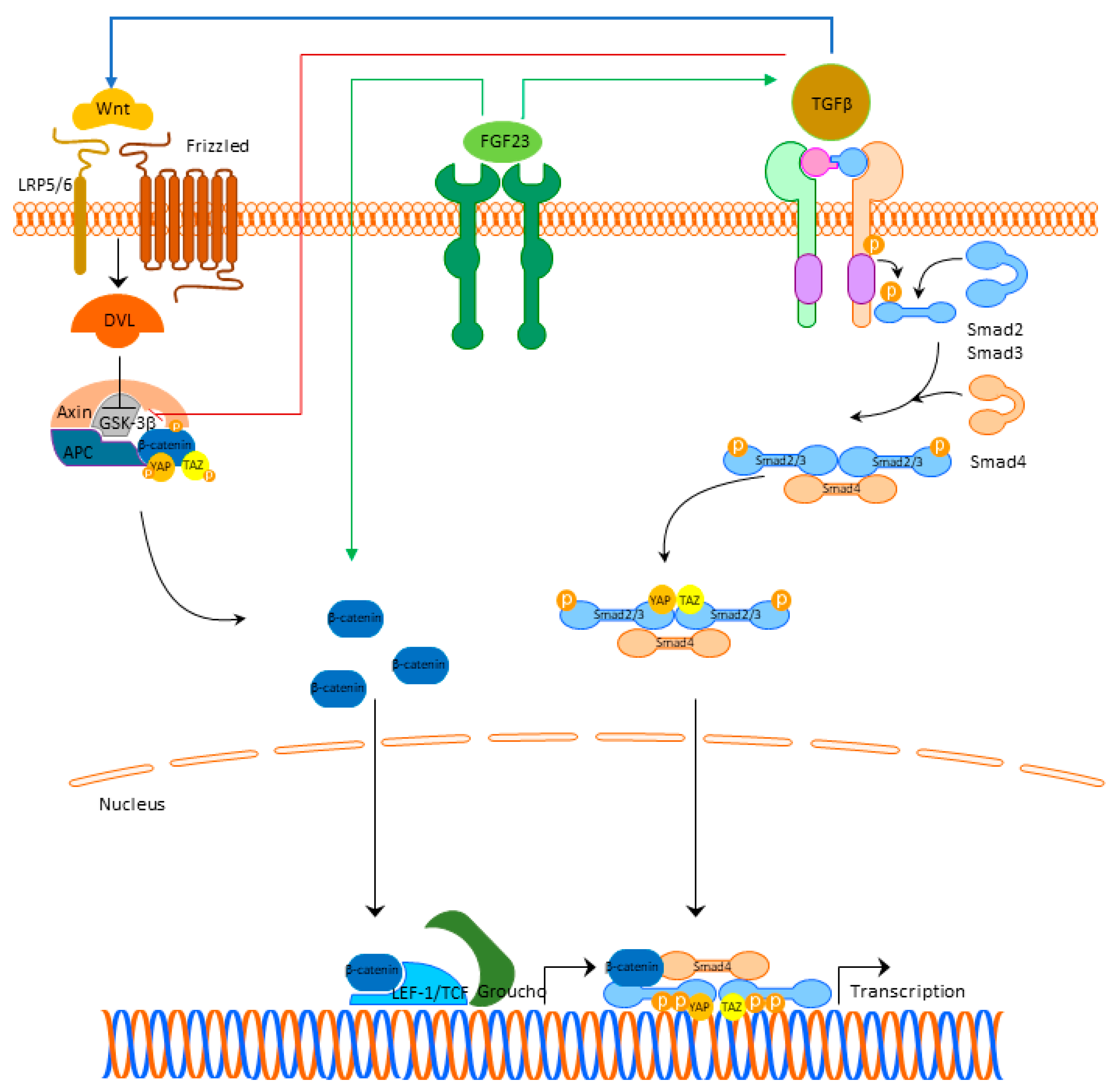 Figure 2. FGF23 signaling and cardiac fibrosis. Abbreviations: FGF23—fibroblast growth factor 23, FGFR4—fibroblast growth factor receptor 4, LRP 5/6—lipoprotein-receptor-related proteins 5 and 6, DLV—Disheveled protein, APC—Adenomatous Polyposis Coli, GSK3β—glycogen synthase kinase 3β, YAP—Yes-associated protein, TAZ—transcriptional coactivator with PDZ-binding domain, LEF/TCF—lymphoid enhancer-binding factor/β-catenin-T-cell factor, TGFβ—transforming growth factor beta.
Figure 2. FGF23 signaling and cardiac fibrosis. Abbreviations: FGF23—fibroblast growth factor 23, FGFR4—fibroblast growth factor receptor 4, LRP 5/6—lipoprotein-receptor-related proteins 5 and 6, DLV—Disheveled protein, APC—Adenomatous Polyposis Coli, GSK3β—glycogen synthase kinase 3β, YAP—Yes-associated protein, TAZ—transcriptional coactivator with PDZ-binding domain, LEF/TCF—lymphoid enhancer-binding factor/β-catenin-T-cell factor, TGFβ—transforming growth factor beta.References
- Kovesdy, C.P.; Quarles, L.D. FGF23 from Bench to Bedside Perspective. Am. J. Physiol. Ren. Physiol. 2016, 310, 1168–1174.
- Bacchetta, J.; Bardet, C.; Prié, D. Physiology of FGF23 and Overview of Genetic Diseases Associated with Renal Phosphate Wasting. Metab. Clin. Exp. 2020, 103, 153865.
- Beck-Nielsen, S.S.; Mughal, Z.; Haffner, D.; Nilsson, O.; Levtchenko, E.; Ariceta, G.; De Lucas Collantes, C.; Schnabel, D.; Jandhyala, R.; Mäkitie, O. FGF23 and Its Role in X-Linked Hypophosphatemia-Related Morbidity. Orphanet J. Rare Dis. 2019, 14, 58.
- Courbebaisse, M.; Lanske, B. Biology of Fibroblast Growth Factor 23: From Physiology to Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a031260.
- Hu, M.C.; Shiizaki, K.; Kuro-O, M.; Moe, O.W. Fibroblast Growth Factor 23 and Klotho: Physiology and Pathophysiology of an Endocrine Network of Mineral Metabolism. Annu. Rev. Physiol. 2013, 75, 503–533.
- Leifheit-Nestler, M.; Haffner, D. Paracrine Effects of FGF23 on the Heart. Front. Endocrinol. 2018, 9, 278.
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of Fibroblast Growth Factor-23 Signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123.
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho Converts Canonical FGF Receptor into a Specific Receptor for FGF23. Nature 2006, 444, 770–774.
- Czaya, B.; Faul, C. The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. Int. J. Mol. Sci. 2019, 20, 4195.
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular Insights into the Klotho-Dependent, Endocrine Mode of Action of Fibroblast Growth Factor 19 Subfamily Members. Mol. Cell. Biol. 2007, 27, 3417–3428.
- Figurek, A.; Spasovski, G.; Popovic-Pejicic, S. FGF23 Level and Intima-Media Thickness Are Elevated From Early Stages of Chronic Kidney Disease. Ther. Apher. Dial. 2018, 22, 40–48.
- Larsson, T.; Nisbeth, U.; Ljunggren, Ö.; Jüppner, H.; Jonsson, K.B. Circulating Concentration of FGF-23 Increases as Renal Function Declines in Patients with Chronic Kidney Disease, but Does Not Change in Response to Variation in Phosphate Intake in Healthy Volunteers. Kidney Int. 2003, 64, 2272–2279.
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Jüppner, H.; Wolf, M. Fibroblast Growth Factor-23 Mitigates Hyperphosphatemia but Accentuates Calcitriol Deficiency in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215.
- Weber, J.R. Left Ventricular Hypertrophy: Its Prevalence, Etiology, and Significance. Clin. Cardiol. 1991, 14, 13–17.
- Leifheit-Nestler, M.; Siemer, R.G.; Flasbart, K.; Richter, B.; Kirchhoff, F.; Ziegler, W.H.; Klintschar, M.; Becker, J.U.; Erbersdobler, A.; Aufricht, C.; et al. Induction of Cardiac FGF23/FGFR4 Expression Is Associated with Left Ventricular Hypertrophy in Patients with Chronic Kidney Disease. Nephrol. Dial. Transplant. 2016, 31, 1088–1099.
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 Is a Novel Regulator of Intracellular Calcium and Cardiac Contractility in Addition to Cardiac Hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873.
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032.
- Grabner, A.; Schramm, K.; Silswal, N.; Hendrix, M.; Yanucil, C.; Czaya, B.; Singh, S.; Wolf, M.; Hermann, S.; Stypmann, J.; et al. FGF23/FGFR4-Mediated Left Ventricular Hypertrophy Is Reversible. Sci. Rep. 2017, 7, 1993.
- Tanaka, S.; Fujita, S.I.; Kizawa, S.; Morita, H.; Ishizaka, N. Association between FGF23, α-Klotho, and Cardiac Abnormalities among Patients with Various Chronic Kidney Disease Stages. PLoS ONE 2016, 11, e0156860.
- Vainikka, S.; Joukov, V.; Wennström, S.; Bergman, M.; Peliccill, P.G.; Alitalot, K. Signal Transduction by Fibroblast Growth Factor Receptor-4 (FGFR-4): Comparison with FGFR-1. J. Biol. Chem. 1994, 269, 18320–18326.
- Han, X.; Cai, C.; Xiao, Z.; Quarles, L.D. FGF23 Induced Left Ventricular Hypertrophy Mediated by FGFR4 Signaling in the Myocardium Is Attenuated by Soluble Klotho in Mice. J. Mol. Cell. Cardiol. 2020, 138, 66–74.
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular Signaling by Fibroblast Growth Factor Receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149.
- Molkentin, J.D. Calcineurin-NFAT Signaling Regulates the Cardiac Hypertrophic Response in Coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475.
- Zhuo, J.L.; Li, X.C. Proximal Nephron. Compr. Physiol. 2013, 3, 1079–1123.
- Vázquez-Sánchez, S.; Poveda, J.; Navarro-García, J.A.; González-Lafuente, L.; Rodríguez-Sánchez, E.; Ruilope, L.M.; Ruiz-Hurtado, G. An Overview of FGF-23 as a Novel Candidate Biomarker of Cardiovascular Risk. Front. Physiol. 2021, 12, 268.
- Gardner, D.G. Natriuretic Peptides: Markers or Modulators of Cardiac Hypertrophy? Trends Endocrinol. Metab. 2003, 14, 411–416.
- Böckmann, I.; Lischka, J.; Richter, B.; Deppe, J.; Rahn, A.; Fischer, D.C.; Heineke, J.; Haffner, D.; Leifheit-Nestler, M. FGF23-Mediated Activation of Local RAAS Promotes Cardiac Hypertrophy and Fibrosis. Int. J. Mol. Sci. 2019, 20, 4634.
- Kuga, K.; Kusakari, Y.; Uesugi, K.; Semba, K.; Urashima, T.; Akaike, T.; Minamisawa, S. Fibrosis Growth Factor 23 Is a Promoting Factor for Cardiac Fibrosis in the Presence of Transforming Growth Factor-Β1. PLoS ONE 2020, 15, e0231905.
- Hao, H.; Li, X.; Li, Q.; Lin, H.; Chen, Z.; Xie, J.; Xuan, W.; Liao, W.; Bin, J.; Huang, X.; et al. FGF23 Promotes Myocardial Fibrosis in Mice through Activation of β-Catenin. Oncotarget 2016, 7, 64649–64664.
- Schumacher, D.; Alampour-Rajabi, S.; Ponomariov, V.; Curaj, A.; Wu, Z.; Staudt, M.; Rusu, M.; Jankowski, V.; Marx, N.; Jankowski, J.; et al. Cardiac FGF23: New Insights into the Role and Function of FGF23 after Acute Myocardial Infarction. Cardiovasc. Pathol. 2019, 40, 47–54.
- Guo, Y.; Xiao, L.; Sun, L.; Liu, F. Wnt/β-Catenin Signaling: A Promising New Target for Fibrosis Diseases. Physiol. Res. 2012, 61, 337–346.
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT Signalling Pathways during Cardiac Fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349.
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
31 May 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No