 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pingfang Yang | + 1819 word(s) | 1819 | 2021-05-17 07:47:57 | | | |
| 2 | Karina Chen | Meta information modification | 1819 | 2021-05-31 05:45:54 | | |
Video Upload Options
Plants are constantly exposed to dynamic environmental conditions due to their sessile nature, which compels their cells to evolve and acquire the ability to change and survive from their endogenous status rapidly. The internal signal transduction ultimately induces modulation of cellular proteins in response to external stimuli (e.g., light or temperature stress). These post translational modifications (PTMs) impact protein’s location, stability and activity, eventually triggering a faster response. Ubiquitylation is one of the most prevalent PTMs, which was originally identified as a modulator of cellular protein turnover and homeostasis.
1. The Ubiquitylation Machinery and Code
Ubiquitylation describes the process of the conjugation of Ub to substrates, which is sequentially catalyzed by a Ub—activating enzyme (E1), a Ub -conjugating enzyme (E2) and a Ub ligase (E3) [1]. Typically, it forms an iso-peptide bond between the C- terminus of Ub and an ε-amino group of a lysine residue of a substrate but it also can be targeted to other amino acids like Cys, Ser and Thr residues [2][3] or a protein’s N-terminus methionine [4]. Glutamic acid (E), aspartic acid (D) and Alanine (A; neutral) were highly enriched around the Kub sites, however, the basophilic residues histidine (H), arginine (R) and lysine (K) were found to be excluded from the adjacent positions [5][6][7]. The flexibility of conjugation dictates the diversity of ubiquitylation. Ub can be attached to a protein at one residue (mono-ub) or multiple residues (multi-ub), more Ub molecules might be added by E2s/E3s and form polymeric chains (poly-Ub, Figure 1) through selective conjugation to its seven lysine residues (K6, K11, K27, K29, K33, K48 and K63) as well as its N-terminal methionine (M1) [8]. This results in distinct structures and functions.
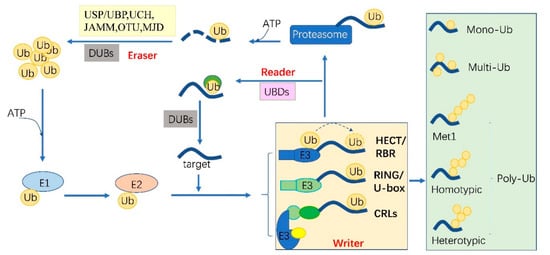
Figure 1. The protein ubiquitylation cascade and its components. Free Ub (Ub) molecules are activated through 3 sequential reactions catalyzed by a Ub-activating enzyme (E1), Ub-conjugating enzyme (E2) and Ub ligase (E3) in an ATP-dependent manner. Based on the transferring mode of Ub Figure 2, the E3s (writer of ubiquitylation) are classed into HECT, RBR, RING, U-box and Cullin-RING E3 ligases. Ub linkage can form into mono-, multimono- (multi-) or poly- ubiquitylation. Ubiquitylation sites are recognized by the proteins carrying Ub binding domains (UBDs, reader of ubiquitylation, including the cap of proteasome) and then direct the targets to be recycled by the deubiquitylases (DUBs, eraser, USP/UBP, UCH, JAMM, OTU and MJD in plants) or 26S proteasome-mediated degradation. HECT, homologous to E6-associated protein C-terminus; RBR, RING-in-between-RING; U-box, a modified RING motif without the full complement of Zn2+-binding ligands; USP/UBP, ubiquitin-specific proteases/ubiquitin-specific processing enzymes; UCH, ubiquitin carboxyl-terminal hydrolases; OUT, ovarian tumor proteases; JAMM, JAB1/MPN/MOV34 domain associated metalloisopeptidase; MJD, Machado-Joseph family proteins.
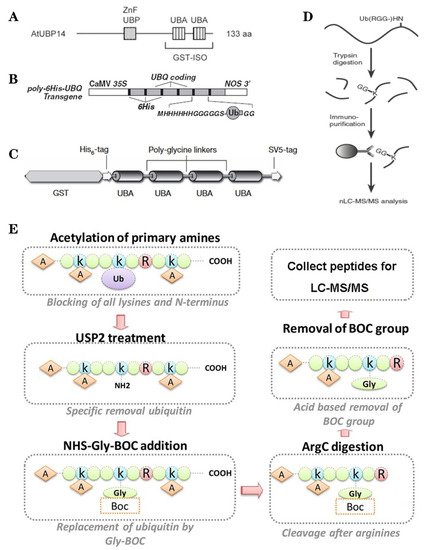
Figure 2. The classic methods of ubiquitylome in plant. (A), a single enrichment step approach using UBA (Ub-associated) motif [9]. (B), tandem affinity purification (TAP) approach using poly His-tag-UBQ (Ub) motif [10]. (C), two-step affinity tandem Ub-binding entities (TUBE) [11]. (D), Affinity chromatography using Lys-ε-Gly-Gly (K-ε-GG) specific antibody [12]. (E), the Ub COFRADIC (combined fractional diagonal chromatography) pipeline [13].
The essential Ub are usually encoded redundantly in the eukaryotic genome with mono-Ub unit fusing to ribosomal or organized as poly-Ub units in a tandem linear [14]. These Ub proteins contain 1–7 Ub units. In Arabidopsis, 12 functional Ub have been identified [15]. These fused- or poly-Ub are initially processed into a single Ub molecule by deubiquitylases (DUBs) before being conjugated to its substrates. The DUBs are also responsible for recycling Ub from the substrates (Figure 1). In plants, there are five different DUB families [16]. Approximately 50 DUBs have been identified in Arabidopsis [16] and 100 of these in human beings [17].
Ubiquitylation and deubiquitylation are tightly regulated in vivo. First, the ATP dependent E1 enzyme captures the Ub through its active-site Cys residue and forms a thioester bond between the C terminus of Ub; then the Ub is transferred onto a Cys residue of E2 [18]. E1 and E2 are relatively conserved in eukaryotes. However, the E3 ligases are diverse among organisms and in different biological processes, where they can selectively recruit specific targets. In humans, there are about 9 E1s and 40 E2s [1] and 600 E3s [17]. These enzymes are more complex in plants. Arabidopsis has 2 E1s and 47 E2s, while approximately 1500 potential E3s; Rice has 6 E1s, 49 E2s and more than 1300 E3s [19][20]. E3 ligases are characterized as ubiquitylation writers with different domains, such as HECT(homologous to E6-associated protein C-terminus), RING, UBOX(a modified RING motif without the full complement of Zn2+-binding ligands), RBR(RING-in-between-RING) and so forth [21]. The ubiquitylated protein can be read by proteins with Ub- binding domains (UBDs, such as Ub-interacting motif (UIMs) and proteasomal receptor) and then be directed to the downstream biological process. Finally, the DUBs or the regulatory cap of the proteasome will erase the Ub from the substrates, thereafter, the free Ub can be recycled [22][23]. The whole cycle constitutes a powerful ubiquitylation language and performs essential signaling functions in all eukaryotes (Figure 1).
2. Methods of Ubiquitylation Detection and Application in Plant Ubiquitylome
The low site-specific stoichiometry, short lifespan, reversible modification, condition-specific expression and complex Ub conjugation architectures bring considerable obstacles in developing deep and accurate catalogs of ubiquitylation. Despite these challenges, greater improvement has been achieved in the identification and verification methods in recent years, especially in plants.
Based on the binding properties between UBDs protein and ubiquitin, three classic methods have been developed to purify the ubiquitylated proteins, including the single-step enrichment, Tandem Affinity Purification (TAP) protocol and two-step affinity tandem Ub binding entities (TUBEs, Figure 2A–C). The single-step enrichment was established to purify the ubiquitylated proteins using affinity matrices through UBDs and the monoclonal anti-ubiquitin antibodies, directly [9]. This method has successfully identified hundreds of ubiquitylated proteins but many false positives were also identified due to non-specific binding under the nondenaturing conditions. TAP protocol greatly avoid this shortcoming, with an initial production of a stable Arabidopsis transgenic line expressing poly-UBQ gene encoding Ub monomers N-terminally tagged with hexahistidine and then purified with sequential Ub-affinity and strong denaturing nickel chelate-affinity chromatography [10]. Saracco et al. (2009) reported that although only 54 non-redundant targets expressed by 90 possible isoforms were identified by mass spectrometry due to the high stringency of TAP, the accuracy was highly improved. Two-step affinity took advantage of the same Arabidopsis transgenic line of TAP, adopting TUBEs developed by Lopitz-Otsoa et al. [24], which drastically improved the purification stringency and yielded about 950 ubiquitylation substrates in the whole Arabidopsis seedlings [25].
The above methods brought great leap for ubiquitylation identification in plants but have not revealed the exact modified site, through site-directed mutagenesis of the lysine residues, therefore, few ubiquitylated sites have been verified [25]. For the lysine ubiquitylation, when the modified protein is digested by trypsin, the remains becomes a specific C-terminal remnant of Lys-ε-Gly-Gly (K-ε-GG, DiGly). Searching spectra for the typical DiGly footprint, Maor et al. (2007) successfully identified 85 precise DiGly footprints on 56 proteins in Arabidopsis [9]. The development of antibodies that recognize DiGly remnant was the first breakthrough that made the proteome-wide investigation of the exact ubiquitylation sites by LC-MS/MS possible (Figure 2D) [12]. Through affinity chromatography with K-ε-GG specific antibody, the tryptic ubiquitylated peptides were efficiently enriched, thereafter, analyzed by high quality MS/MS, which paved a way for real ubquitylome and as a result, thousands of proteins were identified in different species [5][26][27]. K-ε-GG antibody can also unbiasedly recognize the epitope on the Ub itself and provide the information for poly-linkage sites. However, the K-ε-GG antibody cannot capture modifications occurring at the N-terminal or other residues, moreover, neither can it differentiate the tryptic cleavage of other small related protein modifiers [28], such as small Ub-related modifier (SUMO).
Ub combined fractional diagonal chromatography (COFRADIC) method was established as a complementary alternative to the K-ε-GG antibody [29]. This protocol first blocks all primary amines (lysines and N termini) via chemical acetylation and removes Ubs with a plant specific DUB USP2cc, then attaches a chemical handle to these free primary amines, subsequently isolating peptides via two consecutive reverse-phase HPLC (RP-HPLC) runs. Afterwards, the handle is removed by ArgC that cleaves the sequence after arginine. As USP2cc specifically recognizes the last five amino acids [30], COFRADIC can successfully avoid the false positive with other Ub-like modifications. USP2cc only recognizes Ub independent of other affected residues, allowing the identification of ubiquitylation on other residues in addition to lysine. Walton et al. (2016) used COFRADIC to identify 16 proteins with N-terminal ubiquitylation in Arabidopsis (Figure 2E) [13]. However, due to the biochemistry-associated bias, COFRADIC generates peptides that are probably too long or too short to be identified by MS/MS and that are difficult for branched Ub chains detection. Therefore, combining the K-ε-GG antibody affinity and the COFRADIC method may provide a deeper insight into ubquitylome.
When a Ub is added to the N-terminal methionine (M) of another Ub, it forms a linear poly-Ub chains that is not detectable by all the above methods. This is because the KGG- antibody does not recognize the characteristic GGMQIFVK peptides while other N-terminally tagged Ub constructs prevent linear poly-Ub chain assembly (e.g., TAP). Kliza et al. (2017) established a new method of identifying linear poly-Ub-modified proteins, in which a lysine-less internally streptavidin tagged Ub (INT-Ub.7KR) was first constructed, followed by stable isotope labeling of amino acids in cell culture (SILAC)-based mass spectrometry [31]. Using this method, several known linear poly-Ub targets were successfully validated in T-REx HEK293T cells, which provided an effective strategy for liner poly-Ub detection. This method could be modified and be applied in plant proteomics research in the future.
Thanks to the remarkable development of the LC-MS/MS for proteome analysis with high sensitivity and resolution, it is now possible to quickly identify and quantify ubiquitylated proteins in high throughput. Precise relative quantification of ubiquitylated peptides and sites is a big challenge. SILAC has been successfully used for comparison of ubiquitylation dynamics in animal cells. However, its application is rather difficult in plants due to the low efficiency of the in vivo protein labelling. The optimized label-free methods have been proven to be effective in some plants such as petunias, rice and maize [5][6][7], although the accuracy and repeatability require further improvements. In plants, comparative proteome, the advanced tag label methods, for example, isobaric tags for relative and absolute quantitation (iTRAQ) or tandem mass tag (TMT), have been widely used but the K-ε-GG antibody cannot recognize the di-Gly remnant when its N-terminus is derivatized with iTRAQ or TMT. Rose et al. (2016) labelled the ubiquitylated proteins with TMT10 after elution with K-ε-GG antibody and 9000 ubiquitylated peptides were quantified using up to 7 mg labelled sample [32]. To improve the sensitivity and throughput, Namrata et al. (2016) recently developed a rapid and multiplexed protocol termed UbiFast, in which the K-ε-GG antibody is first labelled with TMT and then used to isolate the ubiquitylated peptides. UbiFast facilitated quantification of 10,000 ubiquitylation sites from only 500 μg peptides, which makes large scale comparative ubquitylome more accurate and sensitive [33].
References
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764.
- Pao, K.-C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; van Aalten, D.M.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 2018, 556, 381–385.
- Wang, X.; Herr, R.A.; Hansen, T.H. Ubiquitination of substrates by esterification. Traffic 2012, 13, 19–24.
- Breitschopf, K.; Bengal, E.; Ziv, T.; Admon, A.; Ciechanover, A. A novel site for ubiquitination: The N-terminal residue and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998, 17, 5964–5973.
- He, D.; Li, M.; Damaris, R.N.; Bu, C.; Xue, J.; Yang, P. Quantitative ubiquitylomics approach for characterizing the dynamic change and extensive modulation of ubiquitylation in rice seed germination. Plant J. 2020, 101, 1440–1447.
- Guo, J.; Liu, J.; Wei, Q.; Wang, R.; Yang, W.; Ma, Y.; Chen, G.; Yu, Y. Proteomes and Ubiquitylomes Analysis Reveals the Involvement of Ubiquitination in Protein Degradation in Petunias. Plant Physiol. 2017, 173, 668–687.
- Wang, Y.-F.; Chao, Q.; Li, Z.; Lu, T.-C.; Zheng, H.-Y.; Zhao, C.-F.; Shen, Z.; Li, X.-H.; Wang, B.-C. Large-scale Identification and Time-course Quantification of Ubiquitylation Events During Maize Seedling De-etiolation. Genom. Proteom. Bioinform. 2020.
- Peng, J.M.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.M.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926.
- Maor, R.; Jones, A.; Nühse, T.S.; Studholme, D.J.; Peck, S.C.; Shirasu, K. Multidimensional protein identification technology (MudPIT) analysis of ubiquitinated proteins in plants. Mol. Cell Proteom. 2007, 6, 601–610.
- Saracco, S.A.; Hansson, M.; Scalf, M.; Walker, J.M.; Smith, L.M.; Vierstra, R.D. Tandem affinity purification and mass spectrometric analysis of ubiquitylated proteins in Arabidopsis. Plant J. 2009, 59, 344–358.
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258.
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 2010, 28, 868.
- Walton, A.; Stes, E.; Cybulski, N.; Van Bel, M.; Inigo, S.; Durand, A.N.; Timmerman, E.; Heyman, J.; Pauwels, L.; De Veylder, L.; et al. It’s Time for Some “Site”-Seeing: Novel Tools to Monitor the Ubiquitin Landscape in Arabidopsis thaliana. Plant Cell 2016, 28, 6–16.
- Callis, J.; Carpenter, T.; Sun, C.-W.; Vierstra, R.D. Structure and evolution of genes encoding polyubiquitin and ubiquitin-like proteins in Arabidopsis thaliana ecotype Columbia. Genetics 1995, 139, 921–939.
- Callis, J. The ubiquitination machinery of the ubiquitin system. Arab. Book Am. Soc. Plant Biol. 2014, 12, e0174.
- Isono, E.; Nagel, M.-K. Deubiquitylating enzymes and their emerging role in plant biology. Front. Plant Sci. 2014, 5, 56.
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 59–70.
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331.
- Du, Z.; Zhou, X.; Li, L.; Su, Z. plantsUPS: A database of plants‘ Ubiquitin Proteasome System. BMC Genom. 2009, 10, 227.
- Stone, S.L.; Hauksdottir, H.; Troy, A.; Herschleb, J.; Kraft, E.; Callis, J. Functional analysis of the RING-type ubiquitin ligase family of Arabidopsis. Plant Physiol. 2005, 137, 13–30.
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 2011, 474, 105–108.
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbe, S. Deubiquitylases from Genes to Organism. Physiol. Rev. 2013, 93, 1289–1315.
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563.
- Lopitz-Otsoa, F.; Rodriguez, M.S.; Aillet, F. Properties of Natural and Artificial Proteins Displaying Multiple Ubiquitin-Binding Domains; Portland Press Ltd.: London, UK, 2010.
- Kim, D.Y.; Scalf, M.; Smith, L.M.; Vierstra, R.D. Advanced proteomic analyses yield a deep catalog of ubiquitylation targets in Arabidopsis. Plant Cell 2013, 25, 1523–1540.
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340.
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A Proteome-wide, Quantitative Survey of In Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol. Cell Proteom. 2011, 10.
- Vierstra, R.D. The expanding universe of ubiquitin and ubiquitin-like modifiers. Plant Physiol. 2012, 160, 2–14.
- Stes, E.; Laga, M.; Walton, A.; Samyn, N.; Timmerman, E.; De Smet, I.; Goormachtig, S.; Gevaert, K. A COFRADIC protocol to study protein ubiquitination. J. Proteome Res. 2014, 13, 3107–3113.
- Renatus, M.; Parrado, S.G.; D’Arcy, A.; Eidhoff, U.; Gerhartz, B.; Hassiepen, U.; Pierrat, B.; Riedl, R.; Vinzenz, D.; Worpenberg, S. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure 2006, 14, 1293–1302.
- Kliza, K.; Taumer, C.; Pinzuti, I.; Franz-Wachtel, M.; Kunzelmann, S.; Stieglitz, B.; Macek, B.; Husnjak, K. Internally tagged ubiquitin: A tool to identify linear polyubiquitin-modified proteins by mass spectrometry. Nat. Methods 2017, 14, 504–512.
- Rose, C.M.; Isasa, M.; Ordureau, A.; Prado, M.A.; Beausoleil, S.A.; Jedrychowski, M.P.; Finley, D.J.; Harper, J.W.; Gygi, S.P. Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst. 2016, 3, 395–403.e394.
- Udeshi, N.D.; Mani, D.C.; Satpathy, S.; Fereshetian, S.; Gasser, J.A.; Svinkina, T.; Olive, M.E.; Ebert, B.L.; Mertins, P.; Carr, S.A. Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat. Commun. 2020, 11, 1–11.


