 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stefan Kittler | + 3236 word(s) | 3236 | 2021-05-26 07:45:12 | | | |
| 2 | Rita Xu | Meta information modification | 3236 | 2021-05-28 08:46:57 | | |
Video Upload Options
Protein L is a surface protein of Peptostreptococcus magnus, showing binding activity against IgGs. Therefore, protein L can be used as affinity ligand in the purification process of antibodies and antibody fragments. Furthermore, analytical tools based on this specific interaction are applied in industry and research.
1. Introduction
In 1986, a new type of drug was approved and introduced to the biopharmaceutical market—the first monoclonal antibody (mAb) with the trade name Orthoclone (OKT3), preventing rejection after kidney transplantation [1]. Ever since, the number of mAbs on the biopharmaceutical market has increased rapidly. Antibodies (Abs) are part of the adaptive immune response and formed by B cells as a response to a specific antigen [2]. They can be divided into five classes (IgG, IgA, IgM, IgD, and IgE), differing in their type and number of heavy chains [3][4][5], specifying their properties and functions. The majority of antibodies consist of at least two identical light and heavy chains, with each chain being subdivided into a constant and variable region (Figure 1) [5][6]. Antibodies bind via noncovalent interaction to antigens and provide a targeted and specific interaction [7][8]. The hypervariable region at the top of the Y structure, called the complementarity-determining region (CDR), is responsible for antigen-specific binding and consists of a light and a heavy chain [4][5][6]. Details about the function of each antibody class and reaction have been extensively discussed previously [2][3][4][5][6][9][10][11][12][13][14][15][16][17].
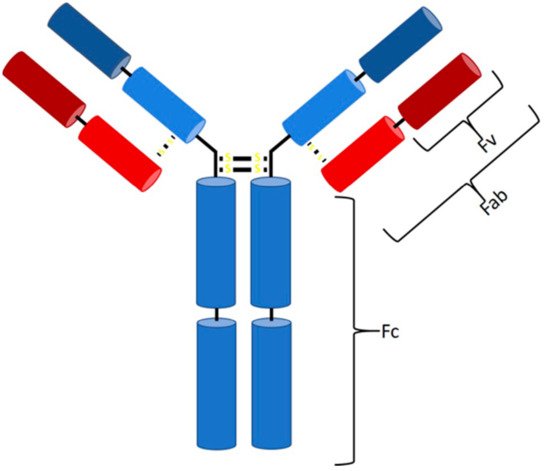
Figure 1. Structure of an IgG antibody. The Fc binding region consists of four heavy chains (2× CH3, 2× CH2), and the Fab region consists of one constant heavy and one constant light chain, while, at the top, two variable chains of each type are located (Fv region).
A wide range of applications for mAbs in therapeutics, biology, biochemistry, and bioanalytics, ranging from drugs against cancer and autoimmune diseases to labeling and detection of virulence factors, has been reported [18][19][20]. They are the largest product class on the biopharmaceutical market today, with a continuous increase in the number of approved products and over 75 currently available mAbs [17][21]. They have an annual market value of around 150 billion dollars, which is approximately 10% of the entire pharmaceutical turnover [21]. Monoclonal antibody production was initially developed by Kohler and Milstein by fusing an antibody-producing B cell and a myeloma cell, leading to the expression of large amounts of identical molecules, so-called monoclonal antibodies [22]. In recent years, mammalian cells lines have emerged as the standard expression host due to their ability to perform posttranslational modifications (PTMs) and extracellular protein production [23][24]. In contrast, microorganisms, such as bacteria and yeast, are not capable of performing human-like glycosylation. However, recent approaches in strain engineering enable human-like glycosylation in microbial hosts [23][25][26]. In contrast to mAbs, microbes are perfectly suited to produce antibody fragments, which do not require glycosylation. Smaller antibody fragments can bind target molecules within the CDR region, which is located in the Fab (fragment antigen binding) region [6][13][17]. However, due to the missing Fc part, these fragments have different pharmacokinetic properties [17][27]. Fragments are no bigger than one-third of a full-length IgG (Fab ~55 kDa), and, as a result, tissues and tumors are penetrated faster, opening a broader field of applications regarding imaging and labeling [28][29][30][31]. On the downside, they show decreased half-life times in the human body [28]. However, this faster clearance can be used as an advantage for the transportation of toxic radioisotopes [28]. Fragments can be subdivided on the basis of the light and heavy chain, namely, Fab (Figure 2a), scFv (Figure 2b), sdAb (Figure 2c), and diabody (Figure 2d), to list the most important ones [32].
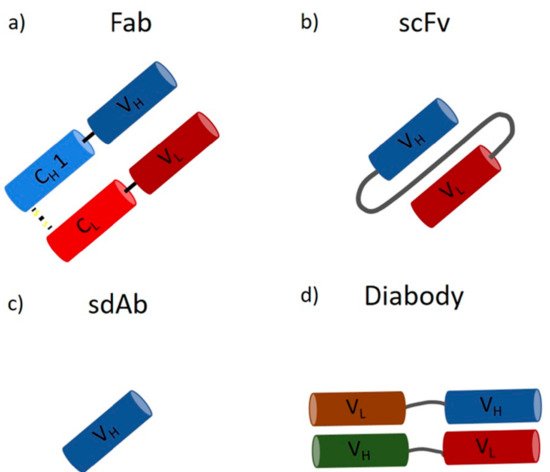
Figure 2. Antibody fragments: (a) the antigen-binding fragment (Fab) contains a variable and a constant domain of a light and heavy chain (size ~55 kDa); (b) the single-chain variable fragment (scFv) consists only of the variable domains, which form the antigen-binding part, linked together by a polypeptidlinker (size ~28 kDa); (c) the single-domain antibody (sdAb) consists of only one variable heavy chain (size ~15 kDa); (d) the diabody consists of two heavy and two light chains forming a bispecific fragment capable of binding two different antigens (size ~50 kDa).
The economic success on the biopharmaceutical market is driven by the advances in the production processes. Mammalian cells are used for the production of mAbs, reaching titers above 5 g/L. Non-glycosylated or non-human-like glycosylated mAbs and antibody fragments are produced mainly in Saccharomyces cerevisiae, Pichia pastoris, and Escherichia coli [23][24]. All production processes require reliable and sensitive process analytical technological tools. The use of real-time monitoring would be beneficial to increase process efficiency and fulfill high-quality requirements [33][34]. Furthermore, recent advances in the productivity have shifted the focus in process development from upstream (USP) toward downstream processing (DSP) [24]. Efficient purification methods are necessary to reach high purity levels, ensuring drug safety. For both applications, affinity ligands can be used. Protein A is currently the state of the art for the purification of mAbs [35]. However, protein A is not applicable for the purification of fragments lacking the Fc region [35].
2. Downstream Processing of mAbs and Fabs
Due to the high market competition and the need to decrease the time to market, it is crucial to determine optimal process conditions leading to high product yields while maintaining the highest quality [13][36]. In Figure 3, an overview of a typical recombinant bioprocess to produce antibody fragments in E. coli is given [24]. In addition to high product yields, strict critical quality attributes (CQAs) have to be reached, which are defined as “a physical, chemical, biological, or microbiological property or characteristic of the product that should be within an appropriate limit, range, or distribution to ensure the desired product quality” [37].
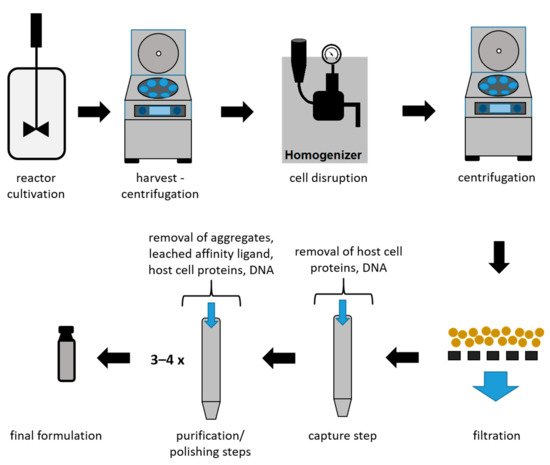
Figure 3. Schematic overview of a bioprocess using E. coli to produce antibody fragments.
As shown in Figure 3, the intracellular protein production in microbial hosts requires several unit operations during the early DSP (harvest to filtration) [23][38]. In the subsequent capture step, large volumes with a low product concentration have to be processed. Furthermore, due to the presence of proteases, short process times are a necessity for this step. Even though a high purity after the capture step is not required, it can be provided using affinity chromatography during the capture step for mAbs and antibody fragments. Therefore, this technique is currently established as the gold standard, achieving not only high recoveries but also high purities. Nevertheless, several chromatography steps are required after the capture step in order to remove host cell proteins (HCPs), DNA, and product-related impurities to reach desired CQAs [39]. A further important reason for subsequent chromatographic steps is the leaching of affinity ligands due to harsh elution conditions [40][41]. The different chromatographic methods for capture and purification/polishing are listed in Table 1, with a focus on the methods used for mAbs and antibody fragments.
Table 1. Comparison of chromatographic methods used for the purification of monoclonal antibodies (mAbs) and antibody fragments. AC (affinity chromatography); AEX (anion exchange chromatography); CEX (cation exchange chromatography); HIC (hydrophobic interaction chromatography); SEC (size-exclusion chromatography); DSP (downstream processing).
| Method | Advantages | Disadvantages | DSP Step | References |
|---|---|---|---|---|
| AC |
|
|
capture | [42][43][44][45] |
| CEX |
|
|
capture (Fc lacking mAbs) purification polishing |
[17][27][44][46][47] |
| AEX |
|
|
purification polishing |
[13][44][46][47][48] |
| HIC |
|
|
polishing | [49][50] |
| SEC |
|
|
polishing | [51] |
To ensure an efficient capturing in the DSP, affinity resins have been established and are mainly used [13][52]. The capability to selectively capture target peptides, while host cell proteins and other molecules bind very weakly or not at all, outperforms other methods [53][54]. Although affinity chromatography is widely used as an initial step, it is expensive and requires harsh elution conditions (pH~3), leading to decreased column stability, ligand leaching, and possible activity loss of the target product [52][55][56]. However, the acidic conditions are an advantage for the required viral inactivation in mammalian production processes [41]. Other chromatographic methods, such as ion exchange (IEX), are mainly used as additional purification steps to remove leached affinity ligands, HCPs, and DNA [41]. These purification and polishing steps can include up to three or four different chromatographic steps to achieve the required product quality (Figure 3) [24].
Nevertheless, affinity resins have emerged as the gold standard for the first step of purification (capture step) of mAbs and antibody fragments; therefore, we explain the most important ligands below.
2.1. Protein A
Protein A originates from the human pathogen Staphylococcus aureus and has a molecular weight of 42 kDa [57][58][59][60]. The protein is anchored to the cell wall and protects the organism by binding IgGs produced by the immune system [59][61]. It contains five homologous binding domains A–E (Figure 4) [40][59][62], and each has the ability to bind IgG subclasses. The S region serves as a signal sequence, and the XM region is used as a cell anchor [56]. The binding domains are composed of three antiparallel alpha helices, and the interaction with the mAbs is primarily based on hydrophobic interaction [55]. In addition to the possibility of binding the Fc region, protein A has shown the ability to bind specific Fab domains [55][59].
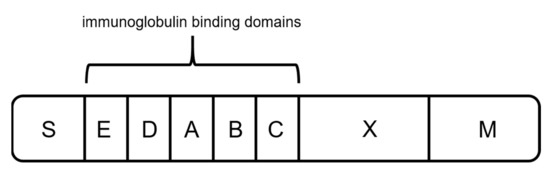
Figure 4. Schematic sketch of protein A, composed of a signal sequence (S) domain, five homologous immunoglobulin binding domains (E, D, A, B, and C), and a cell-wall anchoring domain (X, M) [56].
Each of the immunoglobulin-binding domains can bind to one Fc domain of an antibody or to the Fab region of VH3 human antibodies [55][57][62][63]. Staphylococcal protein A (SPA) binds strongly to Fc domains of IgG1, IgG2, and IgG4 and has a weaker ability to bind IgG3 [55][56][60]. Protein A is used for labeling and purification of mAbs [62], as well as indirect coating for enzyme-linked immunosorbent assays (ELISA) and other immunobinding assays. In nature, protein A makes only 1.7% of the total protein content of Staphylococcus aureus [64]. Since the approval of the first immunosorbent adsorption column by the FDA in 1998 for the therapy of autoimmune diseases, recombinant expression hosts, such as E. coli or Pichia pastoris, have been used for production [64]. Protein A affinity resins have emerged as the primary purification step (capturing) in mAb production processes [40][55]. The DSP of, e.g., HerceptinTM, RituxanTM, MabCampathTM, RemicadeTM, and SimulectTM includes a capture step using protein A affinity chromatography, already resulting in a purity of around 90% after this first chromatographic capture step [13].
2.2. Protein G
Protein G originates from the group G streptococci and has a molecular mass of around 65 kDa [65]. It is the second most used capture ligand in the DSP of mAbs and antibody fragments [55]. Protein G consists of a signal peptide, a cell-wall anchoring domain, and two different binding regions: one located at the N terminus, binding serum albumin, and the second one at the C terminus, interacting with immunoglobulins (Figure 5) [55][65]. For the purification of mAbs, recombinant Protein G is expressed lacking the serum albumin-binding region since albumin would be a contaminant in the formulation of mAbs as biopharmaceuticals [55][66].
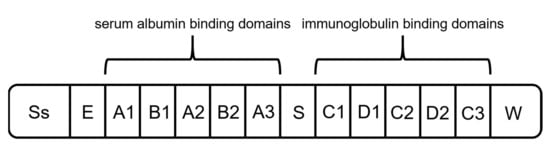
Figure 5. Schematic structure of protein G. It is composed of a signal peptide (Ss), an alanine-rich region (E), an albumin-binding site, an immunoglobulin-binding site, and a cell-wall anchoring region (W) [65].
Protein G interacts with the Fc region of immunoglobulins and binds via hydrogen bonds and salt bridges [55][66][67]. Additionally, protein G shows the capability of binding Fabs through the CH1 domain of IgG1,3,4. Nevertheless, due to the low affinity, purification of fragments is not applicable [55][60][68][69]. Therefore, protein G is mainly used for processes where protein A proves to be unsuitable, e.g., the purification of IgG3. Due to a decreased stability and harsher elution conditions compared to protein A, the number of processes using protein G is considerably lower [55][56][70].
2.3. Protein L
As another alternative to previously mentioned capture ligands, protein L can be used for the binding of mAbs and fragments. Protein L was first isolated in 1985 by Myhre and Erntell as a surface protein of Peptostreptococcus magnus, showing binding activity against IgGs [71][72][73]. The native protein has a size between 76 to 106 kDa, depending on the number of B domains [27][74]. It consists of a signal sequence domain (SS), up to five binding domains (B1–B5), a short spacer region (S), two C repeats with an unknown function, and the wall anchor domain (W) with the transmembrane region (M) (Figure 6) [71]. It was shown that the fifth B domain slightly increases the affinity constant for interaction with kappa light chains (1.5 × 109 M−1 to 2–3 × 109 M−1) [71][75]. Therefore, different versions of recombinant produced protein L consisting of either four or five binding domains are available.
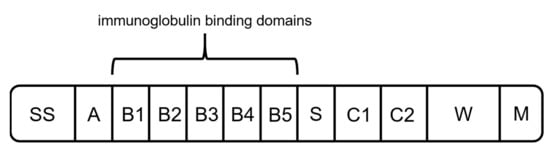
Figure 6. Schematic structure of protein L. It is composed of a hydrophobic signal sequence (SS), the NH2 terminal domain (A), repeated immunoglobulin-binding domains (B1–B5), a spacer region (S), C1 and C2 with unknown function, and the cell-wall anchoring domain [71].
All five binding domains (B1–B5) have a similar structure of an alpha helix and a beta sheet formed by four beta strands [55][76]. Unlike proteins A and G, protein L interacts with the VL domain of kappa light chains [55][71]. It binds to kappa subtypes 1, 3, and 4, enabling the interaction with various types of antibody fragments [77][78]. Furthermore, compared to previously mentioned affinity ligands A and G, protein L binds to a wider range of Ig classes [27][73].
2.4. Comparison of Proteins A, G, and L
Protein A is the most common affinity ligand in the DSP of mAbs and has been investigated in numerous studies in the past years [27]. Protein G is only used in cases where protein A cannot be used, while protein L has only been established for the purification of fragments so far. A low price compared to the other affinity ligands and the suitability for common antibody types make protein A applicable for most capture steps in production processes of mAbs. As depicted in Figure 7, there is a huge price difference between proteins A, G, and L. This is caused by a decreased number of current applications for proteins G and L. Protein L is by far the most expensive ligand. To our knowledge there has been no publication about the large-scale production of protein L to date. Tocaj et al. produced recombinant protein L with four B domains using E. coli on a small scale, obtaining a concentration of 360 mg/L [79]. Nevertheless, recombinant versions produced in E. coli can be purchased from different vendors (Figure 7). The commercial proteins differ in the number of B domains, either four or five, and combinations with affinity tags are available.
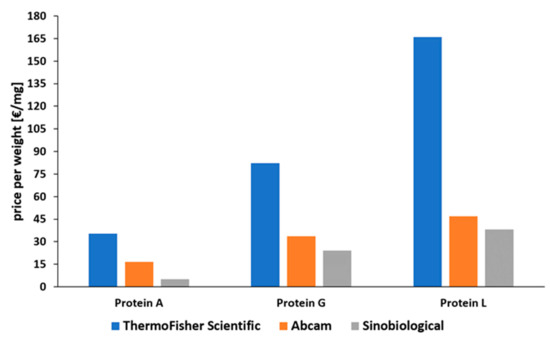
Figure 7. Comparison of prices for recombinantly produced proteins A, G, and L, normalized to EUR per mg [80][81][82]. Status: May 2021.
However, antibody fragments do not have an Fc domain, reducing the applicability of protein A [83][84]. Due to the rising applications and research regarding therapeutic antibody fragments, a modular capture step applicable for both mAbs and fragments is desirable—something that can be provided by protein L [27]. There is no doubt that protein L, due to its unique interaction with kappa light chains, has a huge market potential in the future. Additionally, it shows a high binding capability for human-derived antibodies and fragments [85]. Further interest in antibody fragments is also caused by the possibility to express these target molecules in microbials, such as E. coli [23]. Production costs are significantly lower, higher cell densities can be reached, and virus inactivation is not necessary [86]. However, applications of protein L are still scarce, and its development and improvement are behind that of other affinity ligands. Furthermore, regulatory challenges addressed for protein A are also applicable for protein L. Ligand leaching and limited lifetime would, thus, also be factors to consider.
However, we believe that, in addition to applications as an affinity ligand for product purification, protein L will also have an important role in analytical applications.
3. Is Protein L the Future?
Recombinant proteins in E. coli are usually located intracellularly [39][87]. However, during cultivation, E. coli cells often get leaky, causing an uncontrolled release and, thus, loss of highly valuable product—such as antibody fragments—into the fermentation broth [39]. Today, there is no online detection method for cell leakiness available. Results are obtained too late, and the product is lost in the fermentation broth. Another important analytical aspect in the recombinant production of antibody fragments in E. coli concerns the DSP. Harvested E. coli cells are disrupted, and the product is released together with proteases and other host-specific proteins, which are then removed by several chromatographic steps [39][87]. Furthermore, during the DSP process train, antibody fragments are usually analyzed offline, leading to great time delay. However, online monitoring and control using process analytical technology (PAT) are requested by Quality-by-design (QbD) principles [34][88]. This is particularly challenging for the production of antibody fragments in E. coli, which need to be analyzed in a complex sample background, limiting the applicability of robust and sensitive analytical methods [89]. Currently, recombinant fragments are quantified and analyzed by time-consuming and expensive offline methods, such as immunoassays (ELISA), high-performance liquid chromatography (HPLC,) mass spectrometry (MS), or Western blot [89]. Even though LC–MS-based techniques are constantly being improved, automatization of these techniques is difficult, and they lack high reproducibility [33][89]. More detailed information on currently available PAT tools can be found in a recent review by Wasalathanthri et al. [33].
The implementation of lateral flow, surface plasmon resonance (SPR), impedance, or electrochemical-based techniques as at-line or online tools in the USP and DSP of mAbs or antibody fragments would be highly beneficial. These methods are already used for the detection of virus particles and antibodies [90][91][92]. The use of affinity ligands, such as protein L, as biorecognition elements would provide a platform technology in this respect (Figure 8). The analyte (e.g., mAb, Fab) binds to the immobilized affinity ligand, causing a change in the readout signal (visual, refractive index, impedance, or resistance). Commercial sensors with immobilized protein L are already available to determine binding kinetics of mAbs/antibody fragments. These chips are used for a label-free analysis using SPR or biolayer interferometry (BLI) to determine kinetics and to quantify mAbs or antibody fragments containing kappa light chains [93][94].
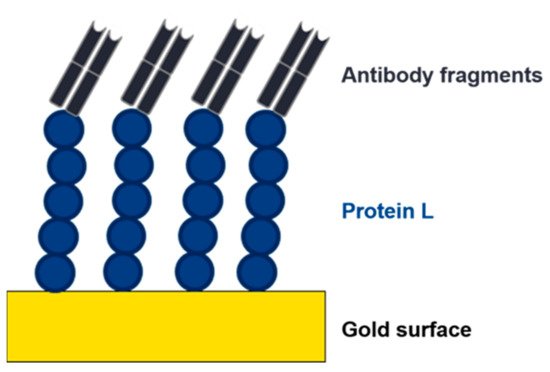
Figure 8. Depiction of an electrochemical impedance spectroscopy (EIS) sensor. An affinity ligand (e.g., protein L) is immobilized to a gold surface. Due to the interaction with an analyte, the charge transfer resistance changes, and the analyte can be detected and quantified.
Protein L is a highly important affinity ligand in these applications due to its interaction with kappa light chains. Antibody fragments and mAbs can be detected with high specificity, which enables universal application in different production processes. Furthermore, protein L shows the highest affinity constants to IgGs derived from human, which is the most important class of mAbs and antibody fragments.
References
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14.
- Grattendick, K.; Pross, S. Immunoglobulins. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–6.
- Conroy, P.J.; Hearty, S.; Leonard, P.; O’Kennedy, R.J. Antibody production, design and use for biosensor-based applications. Semin. Cell Dev. Biol. 2009, 20, 10–26.
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Adaptive Immune Responses to Infection. In Fenner and White’s Medical Virology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 65–76.
- Hnasko, R.M. The Biochemical Properties of Antibodies and Their Fragments. In Methods in Molecular Biology; Metzler, J.B., Ed.; Humana Press: New York, NY, USA, 2015; Volume 1318, pp. 1–14.
- Nelson, A.L. Antibody fragments. In mAbs; Taylor & Francis: Oxfordshire, UK, 2010; Volume 2, pp. 77–83.
- Esela-Culang, I.; Ekunik, V.; Eofran, Y. The Structural Basis of Antibody-Antigen Recognition. Front. Immunol. 2013, 4, 302.
- Wang, H.; Shen, G.; Yu, R. Aspects of recent development of immunosensors. Electrochem. Biosens. 2008, 237–260.
- Schroeder, H.W.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52.
- Gould, H.J.; Beavil, R.L. IgE. In Encyclopedia of Immunology; Elsevier: Amsterdam, The Netherlands, 1998; pp. 1202–1208.
- Edholm, E.-S.; Bengten, E.; Wilson, M. Insights into the function of IgD. Dev. Comp. Immunol. 2011, 35, 1309–1316.
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J. Pathol. 2005, 208, 270–282.
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. Process. Intensif. 2005, 44, 1123–1137.
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520.
- Painter, R.H. IgG. In Encyclopedia of Immunology; Elsevier: Amsterdam, The Netherlands, 1998; pp. 1208–1211.
- Rispens, T.; Vidarsson, G. Human IgG Subclasses. In Antibody Fc; Elsevier: Amsterdam, The Netherlands, 2014; pp. 159–177.
- Bates, A.; Power, C.A. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies 2019, 8, 28.
- Grodzki, A.C.; Berenstein, E. Introduction to the Purification of Antibodies. In Immunocytochemical Methods and Protocols; Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2010; Volume 588, pp. 11–13.
- Andrew, S.M.; Titus, J.A. Purification of Immunoglobulin G. In Current Protocols in Immunology; Wiley: Hoboken, NJ, USA, 2001.
- Miller, L.G.; Goldstein, G.; Murphy, M.; Ginns, L.C. Reversible Alterations in Immunoregulatory T Cells in Smoking. Chest 1982, 82, 526–529.
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30.
- Tomita, M.; Tsumoto, K. Hybridoma technologies for antibody production. Immunotherapy 2011, 3, 371–380.
- Spadiut, O.; Capone, S.; Krainer, F.; Glieder, A.; Herwig, C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014, 32, 54–60.
- Shukla, A.A.; Thömmes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010, 28, 253–261.
- Gerngross, T.U. Advances in the production of human therapeutic proteins in yeasts and filamentous fungi. Nat. Biotechnol. 2004, 22, 1409–1414.
- Gerngross, T. Production of Complex Human Glycoproteins in Yeast. Adv. Exp. Med. Biol. 2005, 564, 139.
- Rodrigo, G.; Gruvegård, M.; Van Alstine, J.M. Antibody Fragments and Their Purification by Protein L Affinity Chromatography. Antibodies 2015, 4, 259–277.
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 2009, 27, 331–337.
- Xenaki, K.T.; Oliveira, S.; Henegouwen, P.M.P.V.B.E. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287.
- Ahamadi-Fesharaki, R.; Fateh, A.; Vaziri, F.; Solgi, G.; Siadat, S.D.; Mahboudi, F.; Rahimi-Jamnani, F. Single-Chain Variable Fragment-Based Bispecific Antibodies: Hitting Two Targets with One Sophisticated Arrow. Mol. Ther. Oncolytics 2019, 14, 38–56.
- Rader, C. Overview on Concepts and Applications of Fab Antibody Fragments. Curr. Protoc. Protein Sci. 2009, 55, 6.9.1–6.9.14.
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136.
- Wasalathanthri, D.P.; Rehmann, M.S.; Song, Y.; Gu, Y.; Mi, L.; Shao, C.; Chemmalil, L.; Lee, J.; Ghose, S.; Borys, M.C.; et al. Technology outlook for real-time quality attribute and process parameter monitoring in biopharmaceutical development—A review. Biotechnol. Bioeng. 2020, 117, 3182–3198.
- Guerra, A.; Von Stosch, M.; Glassey, J. Toward biotherapeutic product real-time quality monitoring. Crit. Rev. Biotechnol. 2019, 39, 289–305.
- Arakawa, T.; Tsumoto, K.; Ejima, D. Alternative downstream processes for production of antibodies and antibody fragments. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 2032–2040.
- Prasnikar, J.; Škerlj, T. New product development process and time-to-market in the generic pharmaceutical industry. Ind. Mark. Manag. 2006, 35, 690–702.
- ICH Guideline Q8 (R2) on Pharmaceutical Development. Available online: (accessed on 13 June 2020).
- Katsuda, T.; Sonoda, H.; Kumada, Y.; Yamaji, H. Production of Antibody Fragments in Escherichia coli. In Antibody Engineering. Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2012; Volume 907, pp. 305–324.
- Wurm, D.J.; Slouka, C.; Bosilj, T.; Herwig, C.; Spadiut, O. How to trigger periplasmic release in recombinant Escherichia coli: A comparative analysis. Eng. Life Sci. 2016, 17, 215–222.
- Ramos-De-La-Peña, A.M.; González-Valdez, J.; Aguilar, O. Protein A chromatography: Challenges and progress in the purification of monoclonal antibodies. J. Sep. Sci. 2019, 42, 1816–1827.
- Shukla, A.A.; Hubbard, B.; Tressel, T.; Guhan, S.; Low, D. Downstream processing of monoclonal antibodies—Application of platform approaches. J. Chromatogr. B 2007, 848, 28–39.
- Mustafaoglu, N.; Kiziltepe, T.; Bilgicer, B. Antibody purification via affinity membrane chromatography method utilizing nucleotide binding site targeting with a small molecule. Analyst 2016, 141, 6571–6582.
- Ayyar, B.V.; Arora, S.; Murphy, C.; O’Kennedy, R. Affinity chromatography as a tool for antibody purification. Methods 2012, 56, 116–129.
- Chahar, D.S.; Ravindran, S.; Pisal, S. Monoclonal antibody purification and its progression to commercial scale. Biologicals 2020, 63, 1–13.
- Ghose, S.; Hubbard, B.; Cramer, S.M. Evaluation and comparison of alternatives to Protein A chromatography: Mimetic and hydrophobic charge induction chromatographic stationary phases. J. Chromatogr. A 2006, 1122, 144–152.
- Fahrner, R.L.; Knudsen, H.L.; Basey, C.D.; Galan, W.; Feuerhelm, D.; Vanderlaan, M.; Blank, G.S. Industrial Purification of Pharmaceutical Antibodies: Development, Operation, and Validation of Chromatography Processes. Biotechnol. Genet. Eng. Rev. 2001, 18, 301–327.
- Ishihara, T.; Yamamoto, S. Optimization of monoclonal antibody purification by ion-exchange chromatography: Application of simple methods with linear gradient elution experimental data. J. Chromatogr. A 2005, 1069, 99–106.
- Grodzki, A.C.; Berenstein, E. Antibody Purification: Ion-Exchange Chromatography. In Immunocytochemical Methods and Protocols; Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2009; Volume 588, pp. 27–32.
- Ghose, S.; Tao, Y.; Conley, L.; Cecchini, D. Purification of monoclonal antibodies by hydrophobic interaction chromatography under no-salt conditions. mAbs 2013, 5, 795–800.
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70.
- Ulmer, N.; Vogg, S.; Müller-Späth, T.; Morbidelli, M. Purification of Human Monoclonal Antibodies and Their Fragments. In Human Monoclonal Antibodies: Methods and Protocols; Steinitz, M., Ed.; Springer: New York, NY, USA, 2019; pp. 163–188.
- Strube, J.; Grote, F.; Ditz, R. Bioprocess Design and Production Technology for the Future. In Biopharmaceutical Production Technology; Wiley: Hoboken, NJ, USA, 2012; pp. 657–705.
- Hage, D.S. Affinity Chromatography: A Review of Clinical Applications. Clin. Chem. 1999, 45, 593–615.
- Ståhl, S.; Kronqvist, N.; Jonsson, A.; Löfblom, J. Affinity proteins and their generation. J. Chem. Technol. Biotechnol. 2012, 88, 25–38.
- Kruljec, N.; Bratkovič, T. Alternative Affinity Ligands for Immunoglobulins. Bioconjugate Chem. 2017, 28, 2009–2030.
- Hober, S.; Nord, K.; Linhult, M. Protein A chromatography for antibody purification. J. Chromatogr. B 2007, 848, 40–47.
- Falugi, F.; Kim, H.K.; Missiakas, D.M.; Schneewind, O. Role of Protein A in the Evasion of Host Adaptive Immune Responses by Staphylococcus aureus. mBio 2013, 4, e00575-13.
- Kobayashi, S.D.; DeLeo, F.R. Staphylococcus aureus Protein A Promotes Immune Suppression. mBio 2013, 4, e00764-13.
- Palmqvist, N.; Foster, T.; Tarkowski, A.; Josefsson, E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb. Pathog. 2002, 33, 239–249.
- Choe, W.; Durgannavar, T.A.; Chung, S.J. Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials 2016, 9, 994.
- Abouelkhair, M.A.; Bemis, D.A.; Kania, S.A. Characterization of recombinant wild-type and nontoxigenic protein A from Staphylococcus pseudintermedius. Virulence 2018, 9, 1050–1061.
- Graille, M.; Stura, E.A.; Corper, A.L.; Sutton, B.J.; Taussig, M.J.; Charbonnier, J.-B.; Silverman, G.J. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA 2000, 97, 5399–5404.
- Romagnani, S.; Giudizi, M.G.; Del Prete, G.; Maggi, E.; Biagiotti, R.; Almerigogna, F.; Ricci, M. Demonstration on protein A of two distinct immunoglobulin-binding sites and their role in the mitogenic activity of Staphylococcus aureus Cowan I on human B cells. J. Immunol. 1982, 129, 596–602.
- Hao, J.; Xu, L.; He, H.; Du, X.; Jia, L. High-level expression of Staphylococcal Protein A in Pichia pastoris and purification and characterization of the recombinant protein. Protein Expr. Purif. 2013, 90, 178–185.
- Sjöbring, U.; Björck, L.; Kastern, W. Streptococcal protein G. Gene structure and protein binding properties. J. Biol. Chem. 1991, 266, 399–405.
- Zhang, H.-C.; Yang, J.; Yang, G.-W.; Wang, X.-J.; Fan, H.-T. Production of recombinant protein G through high-density fermentation of engineered bacteria as well as purification. Mol. Med. Rep. 2015, 12, 3132–3138.
- Jha, R.K.; Gaiotto, T.; Bradbury, A.R.; Strauss, C.E. An improved Protein G with higher affinity for human/rabbit IgG Fc domains exploiting a computationally designed polar network. Protein Eng. Des. Sel. 2014, 27, 127–134.
- Derrick, J.P.; Wigley, D.B. Crystal structure of a streptococcal protein G domain bound to an Fab fragment. Nat. Cell Biol. 1992, 359, 752–754.
- Nilson, B.H.; Lögdberg, L.; Kastern, W.; Björck, L.; Åkerström, B. Purification of antibodies using protein L-binding framework structures in the light chain variable domain. J. Immunol. Methods 1993, 164, 33–40.
- Page, M.; Thorpe, R. Purification of IgG Using Protein A or Protein G. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 2009; pp. 1761–1763.
- Kastern, W.; Sjöbring, U.; Björck, L. Structure of peptostreptococcal protein L and identification of a repeated immunoglobulin light chain-binding domain. J. Biol. Chem. 1992, 267, 12820–12825.
- Myhre, E.B.; Erntell, M. A non-immune interaction between the light chain of human immunoglobulin and a surface component of a Peptococcus magnus strain. Mol. Immunol. 1985, 22, 879–885.
- Darcy, E.; Leonard, P.; Fitzgerald, J.; Danaher, M.; O’Kennedy, R. Purification of Antibodies Using Affinity Chromatography. In Protein Chromatography; Methods in Molecular Biology (Methods and Protocols); Humana Press: New York, NY, USA, 2011; pp. 369–382.
- Graille, M.; Stura, E.A.; Housden, N.G.; Beckingham, J.A.; Bottomley, S.P.; Beale, D.; Taussig, M.J.; Sutton, B.J.; Gore, M.G.; Charbonnier, J.-B. Complex between Peptostreptococcus magnus Protein L and a Human Antibody Reveals Structural Convergence in the Interaction Modes of Fab Binding Proteins. Structure 2001, 9, 679–687.
- Akerström, B.; Björck, L. Protein L: An immunoglobulin light chain-binding bacterial protein. Characterization of binding and physicochemical properties. J. Biol. Chem. 1989, 264, 19740–19746.
- Wikstroem, M.; Drakenberg, T.; Forsen, S.; Sjoebring, U.; Bjoerck, L. Three-dimensional solution structure of an immunoglobulin light chain-binding domain of protein L. Comparison with the IgG-binding domains of protein G. Biochemistry 1994, 33, 14011–14017.
- Nilson, B.H.; Solomon, A.; Björck, L.; Akerström, B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J. Biol. Chem. 1992, 267, 2234–2239.
- Griep, R.; McDougall, J. Analysis and Purification of Antibody Fragments Using Protein A, Protein G, and Protein L. Adv. Struct. Saf. Stud. 2010, 301–315.
- Tocaj, A.; Sjöbring, U.; Björck, L.; Holst, O. High level expression of protein L, an immunoglobulin-binding protein, in Escherichia coli. J. Ferment. Bioeng. 1995, 80, 1–5.
- ThermoScientific Fisher. Available online: (accessed on 10 December 2020).
- Abcam. Available online: (accessed on 10 December 2020).
- Sinobiological. Available online: (accessed on 10 December 2020).
- Chateau, M.; Nilson, B.H.K.; Erntell, M.; Myhre, E.; Magnusson, C.G.M.; Åkerström, B.; Björck, L. On the Interaction between Protein L and Immunoglobulins of Various Mammalian Species. Scand. J. Immunol. 1993, 37, 399–405.
- Paloni, M.; Cavallotti, C. Molecular Modeling of the Interaction of Protein L with Antibodies. ACS Omega 2017, 2, 6464–6472.
- Sheng, S.; Kong, F. Separation of antigens and antibodies by immunoaffinity chromatography. Pharm. Biol. 2012, 50, 1038–1044.
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172.
- Persson, T.; Eckersten, H.; Elen, O.; Hjelkrem, A.-G.R.; Markgren, J.; Söderström, M.; Börjesson, T. Predicting deoxynivalenol in oats under conditions representing Scandinavian production regions. Food Addit. Contam. Part A 2017, 34, 1026–1038.
- Su, Q.; Ganesh, S.; Moreno, M.; Bommireddy, Y.; Gonzalez, M.; Reklaitis, G.V.; Nagy, Z.K. A perspective on Quality-by-Control (QbC) in pharmaceutical continuous manufacturing. Comput. Chem. Eng. 2019, 125, 216–231.
- Gundinger, T.; Pansy, A.; Spadiut, O. A sensitive and robust HPLC method to quantify recombinant antibody fragments in E. coli crude cell lysate. J. Chromatogr. B 2018, 1083, 242–248.
- Nguyen, B.T.; Koh, G.; Lim, H.S.; Chua, A.J.S.; Ng, M.M.L.; Toh, C.-S. Membrane-Based Electrochemical Nanobiosensor for the Detection of Virus. Anal. Chem. 2009, 81, 7226–7234.
- Yu, H.; Kim, K.; Ma, K.; Lee, W.; Choi, J.-W.; Yun, C.-O.; Kim, D. Enhanced detection of virus particles by nanoisland-based localized surface plasmon resonance. Biosens. Bioelectron. 2013, 41, 249–255.
- Laghrib, F.; Saqrane, S.; El Bouabi, Y.; Farahi, A.; Bakasse, M.; Lahrich, S.; El Mhammedi, M. Current progress on COVID-19 related to biosensing technologies: New opportunity for detection and monitoring of viruses. Microchem. J. 2021, 160, 105606.
- Douzi, B. Protein–Protein Interactions: Surface Plasmon Resonance. In Bacterial Protein Secretion Systems: Methods and Protocols; Journet, L., Cascales, E., Eds.; Humana Press: New York, NY, USA, 2017; pp. 257–275.
- Sultana, A.; Lee, J.E. Measuring Protein-Protein and Protein-Nucleic Acid Interactions by Biolayer Interferometry. Curr. Protoc. Protein Sci. 2015, 79, 19.25.1–19.25.26.


