 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stefano Lettieri | + 809 word(s) | 809 | 2021-04-16 05:14:08 | | | |
| 2 | Vicky Zhou | Meta information modification | 809 | 2021-05-26 10:50:02 | | | | |
| 3 | Vicky Zhou | Meta information modification | 809 | 2021-05-26 10:50:25 | | |
Video Upload Options
Photocatalysts indicates the acceleration of a chemical reaction occurring on the surface of a semiconductor material (the catalyst) caused by the presence of light. Illumination of the catalyst causes the generation of free changes (electrons and holes) that can partecipate to chemical reactions, thus causing (or acceleratin) them. Photocatalysis con be used in hydrogen (H2) production (e.g., via water splitting or photo-reforming of organic substrates), CO2 reduction, pollution mitigation and water or air remediation via oxidation (photodegradation) of pollutants. Titanium dioxide (TiO2) is a “benchmark” photocatalyst, thanks to many favorable characteristics.
1. Introduction
A photocatalyst is any material that exhibits photocatalytic properties, i.e., the ability to foster and accelerate specific chemical reactions upon stimulation by light of suitable wavelength. The photocatalytic processes occur via the participation of free charges generated in the photocatalyst through quantum-mechanical transition of electrons into a mobile state via annihilation of the absorbed photons. As these free charges that diffuse toward the materials surface, they can then initiate redox reaction between reactants adsorbed at the photocatalyst surface. Hence, the “suitable” wavelengths mentioned earlier refers to wavelengths which are efficiently absorbed by the photocatalyst.
A material in which photogenerated free charge carriers (also “free carriers” for brevity) are present is not in a thermodynamically stable state: the photogenerated charges will remain in the excited (and mobile) state only for a transient amount of time (carrier lifetime) whose value ultimately depend on the probabilities associated to the various quantum-mechanical decay processes, including all possible non-radiative processes and spontaneous emission (i.e., radiative recombination or luminescence). Longer lifetimes imply greater probability that the free carrier will go into contact with the reagents and (possibly) foster a chemical reaction. From that it follows that conductors (metals) are not suitable as photocatalysts, due to their remarkably short lifetime associated to infra-band electron–electron scattering. Instead, photocatalysts of practical interest are typically semiconductors and, as a consequence, the light-induced free charge carriers involved in fostering redox reactions are electrons and holes belonging to the conduction and valence band (respectively) of the semiconductor.
The photocatalytic action involves conduction band electrons as reactant-reducing agents and valance holes as reactant-oxidizing agents, as schematically shown in Figure 1 for a generic semiconductor. The efficiency of a specific redox reaction will depend on the relative values of the conduction (or valence) band energy of the semiconductor and the reduction (or oxidation) electrode potential of the specific half-reaction, as shown schematically in the caption of Figure 1.
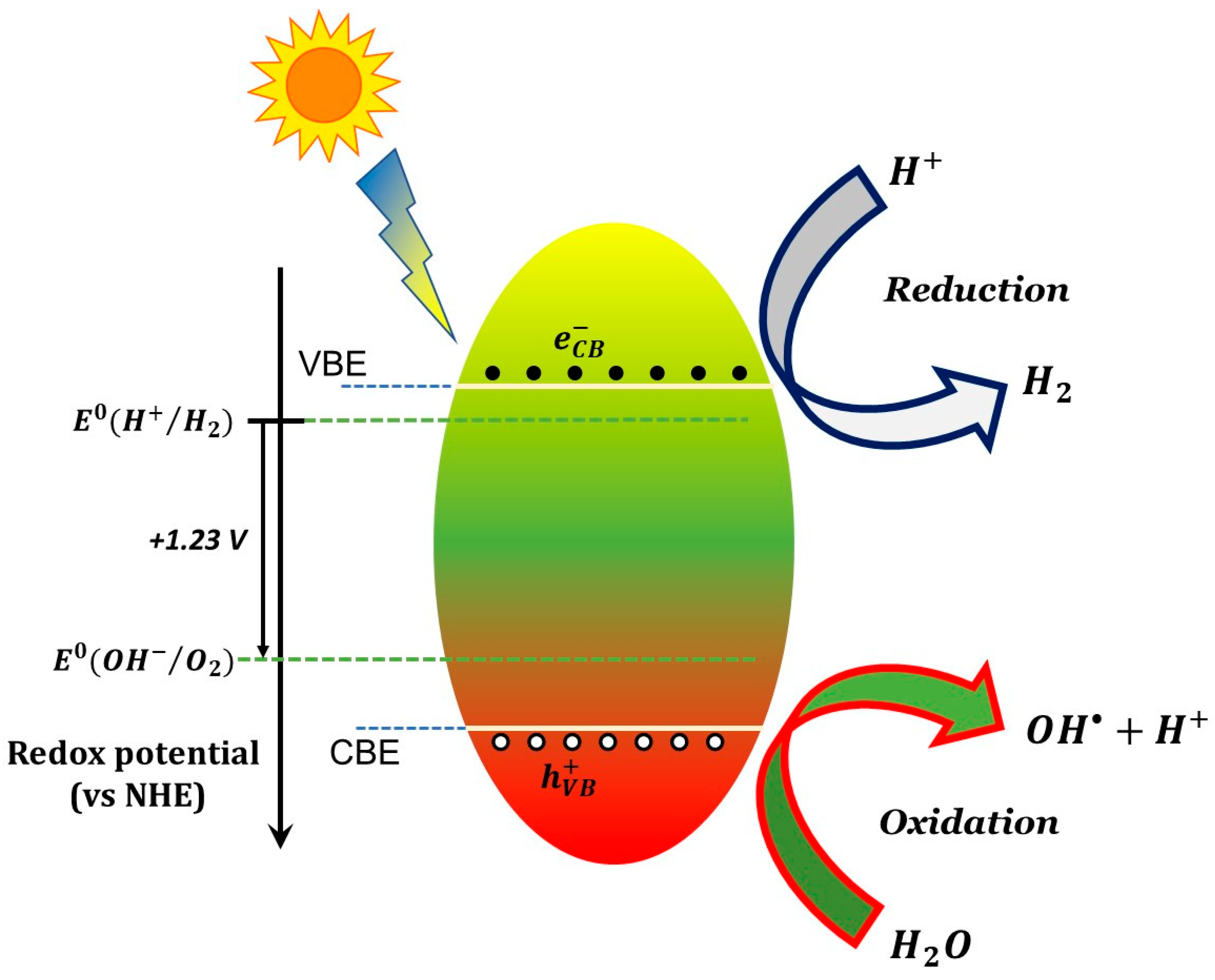
Figure 1. Elementary scheme for a basic photocatalytic process (here, water splitting) caused by illumination of a semiconductor photocatalyst. The scheme shows the reduction of H+ through electrons photogenerated in the conduction band and the oxidation of hydroxyl ions with formation of hydroxyl radicals through photogenerated holes in the valence band. In order for the two processes to occur the electrostatic potential associated to the conduction band edge (CBE) and valence band edge (VBE) have to be lower and greater (respectively) than the electrode potential of H+→H2 and OH−→O2 semi-reactions. The two half-reaction potential are separated by 1.23 Volts and thus a viable photocatalyst for water splitting shall have an energy gap of at least 1.23 eV. Larger gaps are actually needed to compensate for various sources of potential losses (“overpotential”). Similar considerations can be made for other redox reactions, using the corresponding electrode potentials.
2. Limitations of Intrinsic TiO2 as Photocatalyst
The present and near-future trends in TiO2 based photocatalytic technologies are fundamentally influenced by the limitations of intrinsic TiO2, the most important being (A): its inability to be activated by visible light (e.g., sunlight), and (B): the scarce charge separation.
We first consider the problem of visible light-induced activation. As mentioned previously, the band gap energy of anatase TiO2 (the most photocatalytically active polymorph) is about 3.2 eV, which—using the relation in Equation (1)—corresponds to photons of wavelength λg=387 nmlying in the near ultraviolet (or “UV-A”) interval of the electromagnetic spectrum. Unfortunately, this implies that TiO2 is scarcely activated by solar radiation, as the near-UV light accounts for just about 5% of the total solar energy on Earth’s surface.
The second point is the scarce charge separation. Larger proximity of electrons and holes imply larger recombination rates and lower lifetimes of the photogenerated charge carriers, so that most of them will recombine before reaching the surface where the photocatalytic processes (such as ROS generation) shall occur.
In order to overcome these problems, several strategies have been scrutinized. Here, we will schematically classify them in two main categories, namely:
(1) Strategies based on engineered TiO2 nanocrystals, that is centered on modifications and/or control of intrinsic properties of TiO2 nanocrystals, such as the stoichiometric composition or the surface termination (“facet engineering”),
(2) Strategies based on the use of heterojunction photocatalysts, i.e., composites materials or in which TiO2 is electronically coupled with a different material acting as cocatalyst.


