 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Snezana Savic | + 4480 word(s) | 4480 | 2021-05-17 08:05:46 | | | |
| 2 | Rita Xu | Meta information modification | 4480 | 2021-05-26 10:08:00 | | |
Video Upload Options
Due to complex interdependent relationships affecting their microstructure, topical semisolid drug formulations face unique obstacles to the development of generics compared to other drug products.
1. Introduction
Topical semisolid drug products are among the oldest medicinal dosage forms known to human civilization, widely used in treating a variety of skin diseases. Despite their importance and long history of use, the innovations in semisolid products generally lag behind other pharmaceutical product classes. Since topical products commonly produce lower revenues, the development of both novel and generic products is hindered by the projected return on investment-related risks [1]. Namely, the pharmaceutical industry is to invest significant resources to demonstrate the quality, efficacy, and safety of any product before the authorities grant its market authorization [2]. Semisolid formulations, such as ointments, creams, and gels, due to an extremely complex microstructure (i.e., the microscale arrangement of matter and state of aggregation), are accompanied by more complicated, interdependent relationships among the structure, properties, manufacturing process, and performance as compared to solid and injectable dosage forms, that increase the potential for variability and number of failure modes [3][4]. Furthermore, topical drug products face unique obstacles to the development of generics compared to other drug products for which the assessment of bioequivalence is amenable to traditional pharmacokinetic methods [1][5].
As the target site of the most topical semisolid formulations is either the skin or the underlying tissue, due to the none or very low measurable amounts of drug in the systemic circulation, traditionally, establishing bioequivalence in most cases has been based on comparative clinical trials, which are time consuming and expensive, but more importantly, often associated with a high degree of variability and low sensitivity in detecting formulation differences [5][6][7][8][9]. In general, clinical trials require the demonstration of bioequivalence of the prospective generic to the reference/comparator drug product, using one or more clinical endpoints and guaranteeing efficacy by establishing superiority of the tested formulation over a placebo [10]. A clinical response to topical drugs is quite variable due to the numerous patho-physiological factors as well as difficulties involved in the standardization of the applied dose [9]. Likewise, in such cases there is no true placebo since the vehicle components also exert some effects, making the primary endpoints of clinical trials more difficult to meet [2]. As a result, despite the enrollment of a large number of patients in clinical trials (n > 500), frequently, the formulation differences cannot be efficiently detected [10]. This represents a substantial challenge to generic manufacturers and an additional cost for the patients [9]. Indeed, in U.S., in the 2011–2015 period, a significant price increase (almost 276%) was observed for topical generic products, while, simultaneously, oral generic drugs demonstrated a price decrease (21%) [1]. In order to improve the patient access to more affordable topical semisolid drug products on the market, demonstration of bioequivalence requires the usage of alternate approaches which are faster, less expensive, more reproducible, and sensitive to differences in topical products [1][9].
In this context, firstly, to optimize the regulatory requirements for the therapeutic equivalence of topical semisolid drug products, pharmaceutical scientists and dermatologists from academia, industry and regulatory agencies, have proposed the Strawman decision tree and the topical drug classification system [9][10]. Both these approaches highlighted the importance of accounting product specificities, including the properties of pharmaceutical form, drug, site of action and indication. The information on qualitative/quantitative composition and microstructure of the semisolid products being compared represents the basis for rational selection of relevant in vitro/in vivo product performance measures for the determination of bioequivalence [5][9][10][11]. As a result, within the last few years, both European and American regulatory authorities have been advancing regulation relevant to topical generic products, accepting different non-clinical, in vitro/in vivo surrogate methods for topical bioequivalence assessment [12]. From 2012, U.S. Food and Drug Administration (FDA) has continuously published non-binding, product-specific guidelines for generic product development, to identify the appropriate methodology for developing drugs and generating evidence needed to support abbreviated new drug application (ANDA) approval [13]. Over the past five years, a number of relevant guidelines were made public, including an in vitro option to establish bioequivalence of topical semisolid drug products [10][11] (Table 1). As outlined in Table 1, specific in vitro tests that should be performed to support a claim of therapeutic equivalence, in lieu of clinical endpoint studies, highly depend on intrinsic complexity of a specific product.
Table 1. FDA non-binding product-specific draft guidelines for topical generic semisolid drug products that contain in vitro option for establishing bioequivalence [13].
| Drug | Semisolid Dosage Form | Qualitative and Quantitative Sameness Evaluation | Physicochemical Characterization | In Vitro Release Testing | In Vitro Skin Permeation Testing | Additional In Vivo Study | Year |
|---|---|---|---|---|---|---|---|
| Acyclovir | Ointment | + | + | + | 2019 | ||
| Acyclovir | Cream | + | + | + | + | 2016 | |
| Bexarotene | Gel | + | + | + | + | 2019 | |
| Ciprofloxacin hydrochloride | Ointment | + | + | + | 2018 | ||
| Clindamycin phosphate | Gel | + | + | + | 2020 | ||
| Clindamycin phosphate and Tretinoin | Gel | + | + | + | 2020 | ||
| Crisaborole | Ointment | + | + | + | + | PK | 2019 |
| Crotamiton | Cream | + | 2016 | ||||
| Dapsone | Gel | + | + | + | + | PK | 2019 |
| Docosanol | Cream | + | + | + | 2017 | ||
| Doxepin hydrochloride | Cream | + | + | + | + | PK | 2019 |
| Gentamicin sulfate | Cream Ointment |
+ | 2017 | ||||
| Hydrocortisone | Cream | + | 2017 | ||||
| Ivermectin | Cream | + | + | + | + | PK | 2019 |
| Lidocaine | Ointment | + | + | 2016 | |||
| Luliconazole | Cream | + | + | + | + | 2018 | |
| Metronidazole | Gel | + | + | + | 2019 | ||
| Metronidazole | Cream | + | + | + | + | 2019 | |
| Nystatin and Triamcinolone acetonide | Cream Ointment |
+ | 2017 | ||||
| Oxymetazoline hydrochloride | Cream | + | + | + | + | 2019 | |
| Ozenoxacin | Cream | + | + | + | + | 2019 | |
| Penciclovir | Cream | + | + | + | + | 2018 | |
| Pimecrolimus | Cream | + | + | + | + | 2019 | |
| Silver sulfadiazine | Cream | + | + | + | 2017 | ||
| Tacrolimus | Ointment | + | + | + | + | 2018 | |
| Tretinoin | Gel | + | + | + | 2020 | ||
| Tretinoin | Cream | + | + | + | CES | 2020 |
+ indicates methods recommended by the guidelines; PK—in vivo pharmacokinetic study in humans; CES—clinical endpoint studies.
On the other hand, in October 2018, European Medicines Agency (EMA) published for public consultation a universal guideline for topical generic product submission entitled Draft Guideline on Quality and Equivalence of Topical Products. Due to the high diversity of topical products, the complex range of skin conditions that should be treated and the variety of patient needs, this guideline does not provide a single procedure, but states that general recommendations should be adopted on a case-by-case basis [14]. Despite the obvious differences in the manner of proposing the recommendations for generic drug development, EMA requirements are generally similar to those of the FDA. Precisely, according to EMA draft guideline, in case of simple semisolid formulations (e.g., gels, ointments), therapeutic equivalence can be extrapolated from the comparative quality data with the relevant comparator medicinal product (extended pharmaceutical equivalence concept). For this purpose, comparative analysis of pharmaceutical form, qualitative and quantitative composition, microstructure/physical properties, product performance and administration should be performed. In case of complex formulations (e.g., multiphase systems) or those comprising excipients that might affect drug bioavailability and performance, an additional biorelevant test, such as permeation kinetics (in vitro skin permeation, tape stripping or pharmacokinetic bioequivalence) or pharmacodynamic (vasoconstriction assay for corticosteroids or tests relevant for antiseptics and anti-infectives) studies, should be employed (equivalence with respect to efficacy concept) [14] (Figure 1).
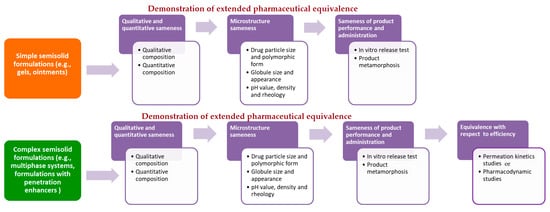
Figure 1. Schematic illustration of the proposed regulatory framework for bioequivalence assessment of topical semisolid drug products according to recently issued EMA draft guideline.
However, it should be noted that the proposed EMA draft guideline is the subject of intensive academia and industry-wide discussions, seeking reliable and robust surrogate bioequivalence methodologies. Despite the significant advances made in the development of generic semisolid products, several limitations have been identified, restricting its successful translation into practice [15].
2. Demonstration of Extended Pharmaceutical Equivalence of Topical Semisolid Drug Products
2.1. Evaluation of Qualitative (Q1) and Quantitative (Q2) Sameness
Drug delivery at the target skin site from topical semisolid products is a complex phenomenon, which depends on the drug physiochemical properties, the disease state and in particular, formulation effects [9]. The formulation composition (excipients’ nature and concentration) is crucial for the therapeutic efficacy, since it directly affects drug solubility and thermodynamic activity, drug release from the dosage form, skin barrier properties and drug penetration/diffusion into/through the skin [16]. Therefore, both European and American regulatory authorities require the demonstration of acceptable Q1 and Q2 sameness, i.e., to document that the test product contains the same excipients in the same quantitative composition as the comparator medicinal product (differences not greater than ±5% are acceptable). According to EMA draft guideline, only excipients whose function is not related to product performance and administration (i.e., antioxidants, preservatives, coloring agents) could be qualitatively and quantitatively different (not more than ±10% is acceptable) [14]. Since the excipients in the comparator product are listed in the patient information leaflet, establishing the Q1 sameness seems to be relatively simple. On the other hand, in order to achieve Q2 sameness, reverse engineering of the comparator product needs to be performed, applying appropriate and validated analytical methods [2][15]. However, due to patent pending or undesirable quality outcome, manufacturers of generic semisolid products are sometimes compelled to modify the formulation composition of the comparator product, and consequently, accomplishing the Q1/Q2 sameness could be a quite challenging task [15][17]. Additionally, as stated in EMA draft guideline, not only formulation composition, but also, the grade of the excipients should be the same, due to its significant impact on the product quality and performance [15][18][19]. For example, analyzing the effect of 6 different petrolatum sources on drug product performance containing petrolatum as the only vehicle, it was observed that diverse grades of petrolatum produced significantly different release rates of a topical steroid, due to variations in the distribution ratios of the hydrocarbons chain lengths [19][20]. However, the grade of excipients used in a comparator product is only available to the regulatory agencies. It is quite demanding to experimentally analyze the grade of any excipient within semisolid formulations, and therefore, assuring the sameness of excipient grades could be difficult to achieve for most generic manufacturers [15].
Although demonstration of Q1/Q2 sameness is considered critical in reducing the failure modes related to the product performance, the variations of key functional excipients, even within the acceptable range (±5%, w/w), can significantly affect the drug bioavailability. In this regard, the results of a recent study performed by Kumar Sharma et al. [21] deserve to be mentioned here, since it investigated the effects of incremental changes in the surfactant concentration (±5%, w/w) on the quality and performance attributes of metronidazole-loaded cream products that meet the definition of Q1/Q2 sameness. Although the monitored quality attributes (globule size, rheology, pH, water activity, rate of drying) practically overlapped, in vitro permeation profiles were remarkably different between the tested formulations. Acceptable 5% w/w change in surfactant concentration obviously led to significant change in the degree of drug saturation during product evaporative metamorphosis, ultimately influencing its performance [21]. This study confirmed that the change in drug thermodynamic activity during metamorphosis, due to slight variations in formulation composition, could significantly alter the drug bioavailability. Although EMA draft guideline asserts that for volatile solvent based topical products, product transformation on administration should be also compared, no methodologies have been proposed for this purpose [14][15]. Therefore, again, the requirement regarding product metamorphosis sameness proves to be difficult to attain. In other words, although different methods have been proposed in the literature (e.g., ATR-FTIR spectroscopy, localized nanothermal analysis and photothermal microspectroscopy combined with multivariate data analysis) [15], there are still limited data on their applicability for the characterization of a wide range of topical semisolid products. Therefore, it is essential that EMA provides more detailed recommendations for studying the product metamorphosis.
2.2. Comparative Characterization of Critical Quality Attributes (CQAs)
Although the criteria for Q1/Q2 sameness are met, due to complex formulation composition and manufacturing process parameters, a generic semisolid product may exhibit differences in the microstructure and arrangement of matter compared to the reference product, that may impact its quality and performance attributes [6][17][22]. Various factors determine specific product microstructure, such as size and shape of dispersed particles, polymorphism, agglomeration, droplet size of the internal phase, excipients’ source/grade, processing conditions and storage [17][22][23]. Therefore, according to the EMA draft guideline, for the demonstration of extended pharmaceutical equivalence, comparative characterization of microstructure/physical properties should be performed, analyzing the CQAs that can influence drug bioavailability, usability or can indicate inconsistency in the manufacturing process and product stability issues. For semisolid formulations, pH value, density, and rheological behavior are identified as the main risk factors that should be closely monitored to gain an assurance of microstructural similarity. For suspension and immiscible phase formulations, additional characterization in terms of drug particle size distribution and polymorphic form, that is, globule size distribution and appearance is required [14]. The similar requirements are set out in the FDA product-specific guidelines containing an in vitro option of bioequivalence assessment. Physicochemical characterization should include comparative analysis of appearance, rheological properties, drug particle size and size distribution, globule size, pH, water activity, and other potentially relevant physical and structure similarity characteristics [11][13]. However, it should be noted that the reliable characterization of microstructure has sparked numerous discussions among different stakeholders (academia, industry and several regulatory agencies) during the last few years. Among other, they imposed the following two questions: (i) which quality attributes are truly critical to the therapeutic performance of topical semisolid dosage forms, as well as (ii) what are the appropriate methodologies for measuring each of these quality attributes without disturbing the original product microstructure [3][4]. Currently, both European and American regulatory authorities do not provide recommendations for the methods that should be utilized for measuring the mentioned CQAs.
Generally, the rheology of semisolid products is highly sensitive to alternations in the product microstructure, and therefore, detailed rheological characterization takes the central role in detection of the potential microstructure differences [22][24]. Furthermore, rheological characterization serves as a useful quality and stability indicator, which could provide additional information concerning batch variability, product sensorial properties (e.g., consistency, spreadability, and feel) and consequently patient compliance [22][25]. Hence, EMA defines specific rheological parameters that should be documented when characterizing the rheological profile of a given formulation. Precisely, (i) a complete flow curve of shear stress (or viscosity) versus shear rate, (ii) yield stress, and (iii) the linear viscoelastic response (storage and loss modulus vs. frequency) should be determined. Additionally, the product’s behavior should be classified according to shear and time effects and described using appropriate metrices (viscosities at specified shear rates across the rheograms (e.g., η100); plastic flow yield stress values; thixotropic relative area (SR); viscoelastic storage and loss moduli (G’ and G”); apparent viscosity; loss tangent (tan δ)) [14]. These parameters should be determined in at least three batches of the test and reference products with at least 12 replicates per batch. In order to declare microstructure equivalence, the 90% confidence interval (CI) for the difference of means of the test and reference products should be included within the acceptance limits of ±10% of the reference product mean, assuming normal distribution of data [14]. This requirement has been intensively disputed in the literature during the last two years as overly restrictive, because it does not take into account the intrinsic variability of topical semisolids [15]. In an attempt to clarify this issue, Pleguezuelos-Villa et al. [23] compared rheological data of Q1/Q2 equivalent test and reference diclofenac diethylamine-loaded emulgels with the results obtained from in vivo pharmacokinetic study in 32 healthy volunteers. Despite statistically significant difference in rheological parameters (90% CI was outside the 90–111% limits), the investigated products could be considered bioequivalent based on the in vivo bioavailability assay. This finding suggests that a difference beyond ±10% between rheological parameters of test and reference products does not necessarily translate into relevant in vivo differences [23]. Similarly, while analyzing the spreadability of three generic formulations that were shown to be equivalent to the innovator product during clinical bioequivalence studies, Kryscio et al. [24] observed that the equivalence in spreadability (inversely proportional to yield stress) is not a prerequisite for product bioequivalence.
In this regard, it should be emphasized that before EMA draft guideline became available for public consultation, all rheological parameters listed above were not a part of routine analysis when releasing new bathes, and therefore, limited data regarding the batch-to-batch variability was available [26]. Hence, Mangas-Sanjuán and coworkers [26] performed comprehensive rheological characterization of 10 batches of a reference product (Daivobet® ointment 50 µg/0.5 mg/g, Leo Pharma A/S, Ballerup, Denmark, containing calcipotriol and betamethasone) to evaluate whether the inter-batch variability of the rheological parameters allows demonstrating equivalence within a ±10% acceptance range. Analyzing the obtained 90% CIs (based on both parametric and non-parametric data analysis), the equivalence for most of the rheological parameters could not be demonstrated. In other words, due to the relatively high inter-batch variability (>10% for several parameters), an acceptance range of ±10% was inappropriate to declare quality equivalence [26]. Generally, the observed high batch-to-batch variability can be derived from the complexity of excipient source (excipient intra-supplier variability), small differences in manufacturing procedure, batch size, storage conditions and aging of the formulations [26][27]. Therefore, in order to overcome the observed limitations of rheological measurements, the authors proposed (i) to widen the acceptance range up to ±20% (which corresponds to those for AUC and Cmax in pharmacokinetic bioequivalence studies) or (ii) to calculate the optimal number of batches required to reach the desired statistical power based on the batch-to-batch variability [26]. Similarly, while characterizing three batches of eight reference blockbuster semisolid drug products in the EU market, Miranda et al. [27] observed that none of the same product batches could be considered as equivalent according to EMA criteria, due to the high variability in rheological parameters (at least two rheological endpoints were statistically different between the batches of the same product). This clearly confirms the need for establishing new microstructure sameness criteria, taking into account the intrinsic variability of the product being studied [15][27]. In this context, Xu and coworkers [28] tried to establish the optimal number of batches and replicates per batch based on different scenarios of inter-batch and intra-batch variability, to accurately demonstrate microstructure similarity between two semisolid products. The calculation of proper sample size is important to disable data manipulation by preventing pharmaceutical companies to choose those product batches that behave similarly. Founded on the simulation-based data analysis, it was concluded that, in cases of low intra- and inter-batch variability, the minimum number of batches should be three, with minimum six units per batch. For the products with up to 5% difference, testing six batches with 12 units per batch or three batches with 24 units per batch could be sufficient to declare equivalence. Finally, in cases when intra- or inter-batch variability exceeds 10%, the number of batches and/or the number of units should be further increased [28].
Additionally, it should be emphasized that usual approach for calculation of CI for the difference of means of the test and reference product, relative to the reference product mean, does not consider the variability in the reference mean estimate [29]. Hence, assuming normal data distribution, Ocaña and collaborators [29] proposed new CI for the test/reference mean ratio, based on the Fieller’s theorem, which takes into account both the within-batch and the between-batch variance, thus enabling more accurate equivalence declaration. Due to the relatively large number of rheological parameters that should be tested as well as high restrictiveness of EMA draft guideline, it was not possible to demonstrate equivalence even between two packaging formats of the same reference product (betamethasone ointment 0.5 mg/g). Hence, for multivariate concepts, such as rheology, Ocaña et al. [29] also suggested to summarize all of the continuous variables to just one or a few variables by means of principal components analysis technique (PCA) (for more details, please see Ocaña et al. [29]). Additionally, several studies noticed that rheological parameters frequently do not follow normal distribution. Therefore, the calculation of 90% CI based on the ratio of geometric means of test and reference products seems to be more appropriate [23][26][29].
On the other hand, from a regulatory point of view, the prerequisite for use of rheology methods as a tool for microstructure characterization of topical semisolids either for quality control or equivalence demonstration is an appropriate standardization of the procedure. However, currently, there are no regulatory recommendations for the standardization, i.e., formal validation of this method. Hence, Simões and coworkers [25] tried to establish a practical approach for validation of the rheological analysis, including the rheometer qualification and the validation of numerous operational critical parameters for a rheology profile acquisition. The experimental results showed that the rheology measurement method can be successfully validated, proving its suitability to determine sameness/differences between the formulations. Likewise, obtained findings inter alia showed that geometry configuration, sample application mode and temperature are critical method variables that should be carefully optimized before each analysis. According to the risk assessment analysis, the thixotropic relative area, oscillatory yield point, flow point, and viscosity related endpoints were defined as highly sensitive and discriminatory monitoring responses [25]. Hence, it is believed that the early inclusion of rheological measurements in product manufacture would allow identifying the factors responsible for microstructure variations, which in turn would assure the satisfying product quality and reduce the overall batch variability [25].
For immiscible phase formulations, such as creams, globule size may directly affect the product stability and performance. Poor control of globule size may result in phase separation, creaming or cracking of the semisolid products [30]. On the other hand, the alterations in globule size among the prospective generic and reference semisolid drug products may impact the amount of drug entrapped in the globule, its partitioning between the oil and water phase, and consequently, drug release and partitioning into the skin [30]. For the given combination of excipients, manufacturing process parameters (e.g., rate of mixing, temperature, order of excipients addition) may significantly impact the globule size [30][31]. All these considerations imply the need for careful monitoring of globule size to ensure the microstructure sameness. However, recent studies imposed several conclusions: (1) globule size can significantly vary from the batch to batch of the same semisolid drug product, (2) differences in globule size do not always correlate with differences in rheology or release profile, and (3) even if EMA criterion for globule size sameness is not fulfilled, two products can still be bioequivalent (as confirmed in human in vivo bioequivalence study) [23][27]. Moreover, it is important to highlight how challenging it may be to analyze the globule size of semisolid products. The characterization of emulsion droplets is usually performed using optical microscopes coupled with appropriate software analysis of the globule size distribution (e.g., using free image-analysis software like Image J, National Institutes of Health, Bethesda, MD, USA), although other techniques have also been proposed (e.g., morphologically directed Raman spectroscopy, laser diffraction) [32]. Generally, the microscopic analysis requires the measurement of thousands of particles to obtain statistically valid particle size distribution [32]. Simultaneously, this analysis is associated with high variability (e.g., coefficient of variation (CV) of almost 38.91% according to Pleguezuelos-Villa et al. [23]) and requires careful standardization of the procedure for sample preparation.
Many failure modes of generic semisolid drug products arise from the differences in the physical and structural properties of the drug compared to the reference product. Generally, the variations in drug particle size, morphology and polymorphic form may affect both bulk qualities (such as rheology, density, content uniformity, and other physical properties) and product performance (such as drug release and efficacy of drug delivery to the target site) [3]. Indeed, recently, it was observed that the size of drug particles was one of the main factors determining acyclovir release from cream formulations [33]. As authors emphasized, particle size of the dispersed acyclovir is the CQA that should be carefully controlled when developing acyclovir topical creams with desired performance characteristics [33]. However, it is quite difficult to ensure the same drug particle size and morphology in the prospective generic product as in the reference product, because they are highly dependent on the properties of the raw drug. Although milling of the raw drug can help reduce the particle size and thus obtain comparable sizes to the reference, the ultimate particle size depends on the solubilization effect of the cosolvents/surfactants used in the formulations and/or the shearing effects during the homogenization process of the creams. On the other hand, unlike drug particle size and morphology that can be relatively easy determined using the microscopic techniques, the characterization of drug-specific polymorph requires more sophisticated techniques like X-ray diffractometry, thermal analysis, or others. It can be technically quite difficult to analyze the polymorphic form in semisolid products, due to the risk of form conversion, including crystallinity change, during the sample preparation [32].
A formulation’s pH value may have considerable influence on drug solubility, ionization state, polymorphic state, ratio of dissolved to undissolved drug, amount of drug in the phase in contact with the skin, as well as a formulation’s viscosity and stability, thus determining the product quality and performance [4]. Likewise, safety and local tolerance of topical semisolid products may be affected by their pH value, since application of a topical formulation with pH that markedly deviates from the skin pH may cause irritation, particularly when accompanied with a skin condition/disease [30]. Considering that the final product’s pH value is governed by the inherent nature of the drug, excipients interactions within the formulation, and also by the manufacturing process (e.g., order of components’ addition) [30][31], it is clear that pH, as a CQA, should also be monitored for the demonstration of extended pharmaceutical equivalence. For example, in acyclovir cream products, the soluble fraction of acyclovir in the aqueous phase has been identified as the critical factor for the product performance and its therapeutic outcome. Since acyclovir has two pKa values (2.27 and 9.25), depending on the pH of the aqueous phase, soluble fraction of acyclovir may be present in cationic, zwitterionic, and anionic forms, which may have different skin permeation potential [7]. In this context, recently, Kamal and coworkers [33] investigated the effects of different formulation variables (propylene glycol, poloxamer and sodium lauryl sulfate concentrations) and different pHs of the aqueous phase (4, 6.5, 9) on critical quality and performance attributes of acyclovir cream. Interestingly, the intentional change in pH of the aqueous phase did not significantly affect acyclovir final concentration in the aqueous phase, and consequently had negligible effect on acyclovir permeation and skin retention. It appears that other excipients involved (predominantly propylene glycol) masked the effect of pH on ionization of acyclovir molecules and their delivery into and through the skin in vitro [33]. Additionally, it should be noted that, while analyzing pH values of three batches of eight reference semisolid drug products, Miranda et al. [27] observed significant inter-batch differences in the pH value, despite the same formulation and processing conditions. Although, undoubtedly, the same composition and microstructure attributes (inter alia pH values) related to the comparator product can help ensure the same therapeutic performance of the prospective generic, both mentioned studies again impose the conclusion that acceptance limit (90% CI within ±10% of the reference product mean) proposed by EMA for pH sameness is too restrictive, i.e., more reasonable criteria should be specified.
Finally, according to EMA draft guideline, comparative analysis of density, as another important quality attribute, should also be performed during microstructure characterization for abridged bioequivalence demonstration. Density of a sample directly affects the dose withdrawn and applied by patients—the lower dose will be drawn from the formulation with lower density compared to high density one [34]. However, unlike rheological properties that have been the subject of various studies during the last few years, literature data whether and how the variations in density of Q1/Q2 equivalent topical semisolid products affect the product performance are still lacking. Consequently, since acceptance criteria for a generic product, according to EMA draft guideline, are ultimately dependent on reference product results [27], detailed investigation of batch-to-batch variability of density is needed.
References
- Kwa, M.C.; Tegtmeyer, K.; Welty, L.J.; Raney, S.G.; Luke, M.C.; Xu, S.; Kong, B. The relationship between the number of available therapeutic options and government payer (medicare part D) spending on topical drug products. Arch. Derm. Res. 2020, 312, 559–565.
- Lenn, J.; Brown, M. Cost-Effective Approaches for Successful Generic Dermal Drug Product Authorization. Available online: (accessed on 18 April 2021).
- Wu, K.; Yeoh, T.; Hsieh, Y.L.; Osborne, D.W. Quality Assessment of API in Semisolid Topical Drug Products. In The Role of Microstructure in Topical Drug Product Development, 1st ed.; Langley, N., Michniak-Kohn, B., Osborne, D.W., Eds.; Springer: Cham, Switzerland, 2019; Volume 36, pp. 109–154.
- Shanley, A. Topical Formulation: Moving from Art to Science. APIs, Excipients, and Manufacturing 2016, Supplement to Pharmaceutical Technology 40 (9). Available online: (accessed on 18 April 2021).
- Raney, S.G.; Franz, T.J.; Lehman, P.A.; Lionberger, R.; Chen, M.L. Pharmacokinetics-based approaches for bioequivalence evaluation of topical dermatological drug products. Clin. Pharm. 2015, 54, 1095–1106.
- Ilić, T.; Pantelić, I.; Lunter, D.; Đorđević, S.; Marković, B.; Ranković, D.; Daniels, R.; Savić, S. Critical quality attributes, in vitro release and correlated in vitro skin permeation-In vivo tape stripping collective data for demonstrating therapeutic (non)equivalence of topical semisolids: A case study of “ready-to-use” vehicles. Int. J. Pharm. 2017, 528, 253–267.
- Krishnaiah, Y.S.; Xu, X.; Rahman, Z.; Yang, Y.; Katragadda, U.; Lionberger, R.; Peters, J.R.; Uhl, K.; Khan, M.A. Development of performance matrix for generic product equivalence of acyclovir topical creams. Int. J. Pharm. 2014, 475, 110–122.
- Mohan, V.; Wairkar, S. Current regulatory scenario and alternative surrogates methods to establish bioequivalence of topical generic products. J. Drug Deliv. Sci. Technol. 2021, 61, 102090.
- Yacobi, A.; Shah, V.P.; Bashaw, E.D.; Benfeldt, E.; Davit, B.; Ganes, D.; Ghosh, T.; Kanfer, I.; Kasting, G.B.; Katz, L.; et al. Current challenges in bioequivalence, quality, and novel assessment technologies for topical products. Pharm. Res. 2014, 31, 837–846.
- Shah, V.P.; Yacobi, A.; Rădulescu, F.Ş.; Miron, D.S.; Lane, M.E. A science based approach to topical drug classification system (TCS). Int. J. Pharm. 2015, 491, 21–25.
- Miranda, M.; Sousa, J.J.; Veiga, F.; Cardoso, C.; Vitorino, C. Bioequivalence of topical generic products. Part 2. Paving the way to a tailored regulatory system. Eur. J. Pharm. Sci. 2018, 122, 264–272.
- Minghetti, P.; Musazzi, U.M.; Casiraghi, A.; Rocco, P. Old active ingredients in new medicinal products: Is the regulatory path coherent with patients’ expectations? Drug Discov. Today 2020, 25, 1337–1347.
- US FDA Product-Specific Guidances for Generic Drug Development. Available online: (accessed on 18 April 2021).
- Committee for Medicinal Products for Human Use (CHMP), EMA. Draft Guideline on Quality and Equivalence of Topical Products, CHMP/QWP/708282/2018. 2018. Available online: (accessed on 18 April 2021).
- Miranda, M.; Cardoso, C.; Vitorino, C. Quality and equivalence of topical products: A critical appraisal. Eur. J. Pharm. Sci. 2020, 148, 105082.
- Simões, A.; Veiga, F.; Vitorino, C.; Figueiras, A. A tutorial for developing a topical cream formulation based on the quality by design approach. J. Pharm. Sci. 2018, 107, 2653–2662.
- Shah, V.P.; Rădulescu, F.Ş.; Miron, D.; Yacobi, A. Commonality between BCS and TCS. Int. J. Pharm. 2016, 509, 35–40.
- Chang, R.K.; Raw, A.; Lionberger, R.; Yu, L. Generic development of topical dermatologic products: Formulation development, process development, and testing of topical dermatologic products. AAPS J. 2013, 15, 41–52.
- Raghavan, L.; Brown, M.; Michniak-Kohn, B.; Ng, S.; Sammeta, S. In vitro release tests as a critical quality attribute in topical product development. In The Role of Microstructure in Topical Drug Product Development, 1st ed.; Langley, N., Michniak-Kohn, B., Osborne, D.W., Eds.; Springer: Cham, Switzerland, 2019; Volume 36, pp. 47–87.
- Baynes, R.; Riviere, J.; Franz, T.; Monteiro-Riviere, N.; Lehman, P.; Peyrou, M.; Toutain, P.L. Challenges obtaining a biowaiver for topical veterinary dosage forms. J. Vet. Pharm. Ther. 2012, 35, 103–114.
- Kumar Sharma, P.; Panda, A.; Parajuli, S.; Badani Prado, R.M.; Kundu, S.; Repka, M.A.; Ureña-Benavides, E.; Narasimha Murthy, S. Effect of surfactant on quality and performance attributes of topical semisolids. Int. J. Pharm. 2021, 596, 120210.
- Rawat, A.; Gupta, S.S.; Kalluri, H.; Lowenborg, M.; Bhatia, K.; Warner, K. Rheological characterization in the development of topical drug products. In The Role of Microstructure in Topical Drug Product Development, 1st ed.; Langley, N., Michniak-Kohn, B., Osborne, D.W., Eds.; Springer: Cham, Switzerland, 2019; Volume 36, pp. 3–45.
- Pleguezuelos-Villa, M.; Merino-Sanjuán, M.; Hernández, M.J.; Nácher, A.; Peris, D.; Hidalgo, I.; Soler, L.; Sallan, M.; Merino, V. Relationship between rheological properties, in vitro release and in vivo equivalency of topical formulations of diclofenac. Int. J. Pharm. 2019, 572, 118755.
- Kryscio, D.R.; Sathe, P.M.; Lionberger, R.; Yu, L.; Bell, M.A.; Jay, M.; Hilt, J.Z. Spreadability measurements to assess structural equivalence (Q3) of topical formulations-a technical note. AAPS Pharm. Sci. Tech. 2008, 9, 84–86.
- Simões, A.; Miranda, M.; Cardoso, C.; Veiga, F.; Vitorino, C. Rheology by design: A regulatory tutorial for analytical method validation. Pharmaceutics 2020, 12, 820.
- Mangas-Sanjuán, V.; Pleguezuelos-Villa, M.; Merino-Sanjuán, M.; Hernández, M.J.; Nácher, A.; García-Arieta, A.; Peris, D.; Hidalgo, I.; Soler, L.; Sallan, M.; et al. Assessment of the inter-batch variability of Microstructure Parameters in Topical Semisolids and Impact on the Demonstration of Equivalence. Pharmaceutics 2019, 11, 503.
- Miranda, M.; Cova, T.; Augusto, C.; Pais, A.A.C.C.; Cardoso, C.; Vitorino, C. Diving into batch-to-batch variability of topical products—A regulatory bottleneck. Pharm. Res. 2020, 37, 218.
- Xu, Z.; Mangas-Sanjuán, V.; Merino-Sanjuán, M.; Merino, V.; García-Arieta, A. Influence of inter- and intra-batch variability on the sample size required for demonstration of equivalent microstructure of semisolid dosage forms. Pharmaceutics 2020, 12, 1159.
- Ocaña, J.; Monleón-Getino, T.; Merino, V.; Peris, D.; Soler, L. Statistical methods for quality equivalence of topical products. 0.5 Mg/g Betamethasone Ointment as a Case-Study. Pharmaceutics 2020, 12, 318.
- Namjoshi, S.; Dabbaghi, M.; Roberts, M.S.; Grice, J.E.; Mohammed, Y. Quality by design: Development of the quality target product profile (QTPP) for semisolid topical products. Pharmaceutics 2020, 12, 287.
- Simões, A.; Veiga, F.; Vitorino, C. Progressing towards the sustainable development of cream formulations. Pharmaceutics 2020, 12, 647.
- Osborne, D.W.; Dahl, K.; Parikh, H. Determination of particle size and microstructure in topical pharmaceuticals. In The Role of Microstructure in Topical Drug Product Development, 1st ed.; Langley, N., Michniak-Kohn, B., Osborne, D.W., Eds.; Springer: Cham, Switzerland, 2019; Volume 36, pp. 89–106.
- Kamal, N.S.; Krishnaiah, Y.S.R.; Xu, X.; Zidan, A.S.; Raney, S.; Cruz, C.N.; Ashraf, M. Identification of critical formulation parameters affecting the in vitro release, permeation, and rheological properties of the acyclovir topical cream. Int. J. Pharm. 2020, 590, 119914.
- Murthy, N.S. Critical Quality Attributes (Q3 Characterization) of Topical Semisolid Products. 2016. Available online: (accessed on 18 April 2021).


