 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | temitope fasanya | + 2053 word(s) | 2053 | 2021-05-12 08:31:15 | | | |
| 2 | Camila Xu | Meta information modification | 2053 | 2021-07-01 03:58:18 | | |
Video Upload Options
The cell cycle is an important cellular process whereby the cell attempts to replicate its genome in an error-free manner.
1. Introduction
The cell cycle is a process through which cells faithfully replicate their genetic material. Strict control over its progression is therefore needed to avoid errors that could result in cell death or cell malignancy. As such, understanding how the cell cycle is affected by external and internal stresses is of the utmost importance, in particular, the stress caused by hypoxia, or reduced oxygen availability. Hypoxia is an important factor in embryo development but is also present in numerous pathological settings such as ischaemic events and cancer [1]. Oxygen is fundamental for both energy homeostasis and cellular viability, therefore, to deal with such stresses, cells possess complex response mechanisms that aim at restoring oxygen homeostasis.
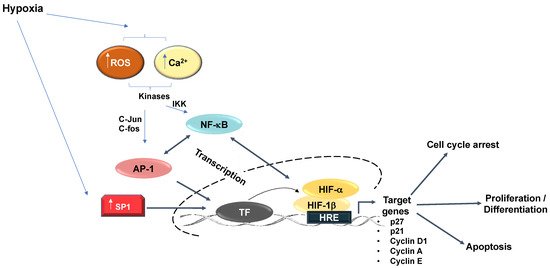
2. Hypoxia Signalling Pathway
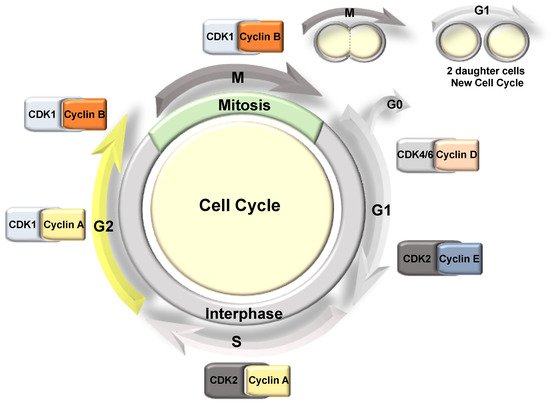
3. Cell Cycle Overview
4. Hypoxia Mediated Transcriptional Effects on Cell Cycle Components
Hypoxia plays a central role in diverse aspects of cancer biology as a hypoxic microenvironment is prevalent in solid tumours. It is known that oxygen levels regulate cell proliferation and that depending on the cell type, hypoxia can inhibit cell proliferation by inducing cell cycle arrest [15][16][17]. However, tumour cells often adapt to survive in such hypoxic conditions. This adaptation is partially promoted by the essential role HIF plays in the transcriptional regulation of genes associated with angiogenesis, apoptosis, cell proliferation, and energy metabolism [2]. HIF-dependent activation of these genes provides tumour cells with an adaptive advantage over normal cells during periods of hypoxic stress.
The Myc transcription factor, which is also associated with cell proliferation, is similarly overexpressed in various cancer types [18]. Myc is known to repress the expression of the CDK cyclin inhibitors p21 and p27 [18]. In hypoxia, the expression of p21 and p27 is induced in a HIF-1α dependent manner, partially by displacing Myc from their promoters [15]. Interestingly, HIF-2α cooperates with Myc and promotes cell proliferation in renal clear cell carcinomas (RCC) and other cell types [19]. In RCC, HIF-2α is also known to induce Cyclin D1 via its binding to an enhancer site [20] (Figure 3). The effects of HIF-2α on cell cycle control in RCC cells have become even clearer following data derived from treatments with a specific HIF-2α inhibitor (PT2399), in which the cell cycle was identified as the main signature controlled by HIF-2α [21][22].
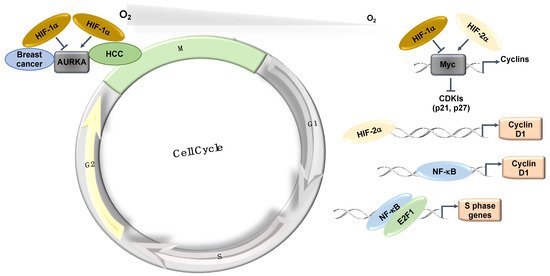
The mitotic kinase—Aurora A (AURKA)—also has an important role in mitotic progression and has been related to oncogenic phenotypes [23]. AURKA is overexpressed in a variety of different tumours and has been implicated in cell transformation and centrosome amplification [24][25]. In hepatocellular carcinomas (HCC), the increased levels of AURKA do not always correlate with gene amplification [26]. Interestingly, AURKA was shown to be induced by hypoxia in a HIF-1α dependent manner in HepG2 cells [27] as HIF-1α binds to the AURKA promoter and upregulates its transcription, leading to an increase in cell proliferation [27]. This study proposed that AURKA might be involved in the hypoxia-induced proliferation of HCC tumours [27] (Figure 3). In this tumour type, hypoxia induction of HIF-1α and AURKA might be involved in promoting HCC proliferation [27]. However, in breast cancer cells (MCF-7, MDA-MB-231, and SK-Br3), microarray analysis in response to hypoxia illustrates that hypoxia downregulates AURKA [28]. In this study, the authors demonstrated that hypoxia downregulated AURKA via a HIF-1α dependent mechanism, suggesting that HIF-1α is a negative regulator of AURKA in breast cancer tumours [28]. These studies highlight the cell-specific nature of HIF’s function in cell cycle regulation.
Cell division cycle-associated protein 2 (CDCA2) has similarly been shown to be a target of the hypoxia pathway [29]. Analyses of publicly available ChIP-Seq and RNA-Seq datasets demonstrate that CDCA2 is induced in hypoxia and is important for the control of proliferation in prostate cancer cells. The intracellular signal transducer protein, SMAD3, binds to the CDCA2 promoter and recruits HIF-1α. HIF-1α/SMAD3 then mediates the transcriptional regulation of CDCA2 in hypoxic tumours, leading to cancer cell proliferation [29].
HIF’s control of the cell cycle is also mediated by its ability to induce a variety of microRNAs and long non-coding RNAs; this aspect was reviewed in [14][30].
Among all known hypoxia-stimulated transcription factors, the importance of NF-κB has been identified in a wide range of signalling systems, alongside its association with many different diseases. In contrast to its well-established role in immune and inflammatory responses, NF-κB activity is also involved in apoptosis, carcinogenic transformation, and cell cycle transition [31]. The NF-κB family consists of five distinct members, which include RelA (p65), RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52), all of which share a conserved Rel homology domain [31]. Hypoxia activation of NF-κB occurs via several mechanisms, although some of the details still require further investigation. Hypoxia has been shown to control NF-κB via the action of PHDs and FIH [32][33][34], and requires the involvement of calcium/calmodulin-dependent kinase II (CAMKII), transforming growth factor kinase 1 (TAK1) and IκB kinase (IKK) [35][36][37].
NF-κB has several target genes identified with a role in cell cycle progression (reviewed in detail [31]). Ultimately, cyclin D1 provides a predominant link between NF-κB and cell cycle progression (Figure 3). Cyclin D1, in association with CDK4 and CDK6, promotes G1/S phase transition through CDK-dependent phosphorylation of retinoblastoma protein (pRB) [38]. This phosphorylation event releases the transcription factor E2F, which is required for the activation of S-phase-specific genes, although several studies have conversely reported that NF-κB can itself induce cell cycle arrest [39]. Overexpression of RelA has been shown to arrest cells at G1/S-phase transition [40]. c-Rel overexpression leads to cell cycle arrest through p53 protein stabilization, an important upstream activator of the CDK-inhibitor, p21 [41]. Additionally, p21 expression can be further increased by the Formin-2 (FMN2) protein, which is a component in the p14ARF tumour suppressor pathway [42]. In this study, FMN2 was shown to increase with hypoxia stimulation via an NF-κB-dependent mechanism [42]. Moreover, several additional studies have shown that NF-κB can be activated by a hypoxic environment [35][36][43][44].
Finally, a physical and functional interaction exists between the IKK/NF-κB signalling pathway and the cell cycle regulatory proteins of the E2F family that controls S-phase entry, which suggests that NF-κB plays a functional role in controlling cell division [45][46]. Indeed, direct phosphorylation of E2F by IKKα and IKKβ resulted in the nuclear accumulation and enhanced DNA binding of the E2F4/p130 repressor complex, which led to the suppression of E2F-responsive gene expression [45]. In contrast, E2F1 and NF-κB interactions were shown to control the timing of cell proliferation [46] (Figure 3). Despite all of these links between NF-κB and cell cycle regulation, a more systematic and mechanistic analysis of how NF-κB controls the cell cycle in hypoxia is needed.
References
- Mole, D.R.; Ratcliffe, P.J. Cellular oxygen sensing in health and disease. Pediatr. Nephrol. 2008, 23, 681–694.
- Rocha, S. Gene regulation under low oxygen: Holding your breath for transcription. Trends Biochem. Sci. 2007, 32, 389–397.
- Wilson, J.W.; Shakir, D.; Batie, M.; Frost, M.; Rocha, S. Oxygen-sensing mechanisms in cells. FEBS J. 2020, 287, 3888–3906.
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64.
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298.
- van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484.
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528.
- Uchida, C. The retinoblastoma protein: Functions beyond the G1-S regulator. Curr. Drug Targets 2012, 13, 1622–1632.
- Palmer, N.; Kaldis, P. Less-well known functions of cyclin/CDK complexes. Semin Cell Dev. Biol. 2020, 107, 54–62.
- Lim, S.H.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093.
- Dang, F.; Nie, L.; Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021, 28, 427–438.
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166.
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115.
- Ortmann, B.; Druker, J.; Rocha, S. Cell cycle progression in response to oxygen levels. Cell Mol. Life Sci. 2014, 71, 3569–3582.
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956.
- Hackenbeck, T.; Knaup, K.X.; Schietke, R.; Schodel, J.; Willam, C.; Wu, X.; Warnecke, C.; Eckardt, K.U.; Wiesener, M.S. HIF-1 or HIF-2 induction is sufficient to achieve cell cycle arrest in NIH3T3 mouse fibroblasts independent from hypoxia. Cell Cycle 2009, 8, 1386–1395.
- Hubbi, M.E.; Semenza, G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am. J. Physiol. Cell Physiol. 2015, 309, C775–C782.
- Garcia-Gutierrez, L.; Delgado, M.D.; Leon, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244.
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347.
- Schodel, J.; Bardella, C.; Sciesielski, L.K.; Brown, J.M.; Pugh, C.W.; Buckle, V.; Tomlinson, I.P.; Ratcliffe, P.J.; Mole, D.R. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat. Genet. 2012, 44, 420–425, S421-422.
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117.
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111.
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937–23954.
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002, 21, 483–492.
- Tatsuka, M.; Sato, S.; Kanda, A.; Miki, T.; Kamata, N.; Kitajima, S.; Kudo, Y.; Takata, T. Oncogenic role of nuclear accumulated Aurora-A. Mol. Carcinog 2009, 48, 810–820.
- Jeng, Y.M.; Peng, S.Y.; Lin, C.Y.; Hsu, H.C. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin. Cancer Res. 2004, 10, 2065–2071.
- Klein, A.; Flugel, D.; Kietzmann, T. Transcriptional regulation of serine/threonine kinase-15 (STK15) expression by hypoxia and HIF-1. Mol. Biol. Cell 2008, 19, 3667–3675.
- Fanale, D.; Bazan, V.; Corsini, L.R.; Caruso, S.; Insalaco, L.; Castiglia, M.; Cicero, G.; Bronte, G.; Russo, A. HIF-1 is involved in the negative regulation of AURKA expression in breast cancer cell lines under hypoxic conditions. Breast Cancer Res. Treat. 2013, 140, 505–517.
- Zhang, Y.; Cheng, Y.; Zhang, Z.; Bai, Z.; Jin, H.; Guo, X.; Huang, X.; Li, M.; Wang, M.; Shu, X.S.; et al. CDCA2 Inhibits Apoptosis and Promotes Cell Proliferation in Prostate Cancer and Is Directly Regulated by HIF-1alpha Pathway. Front. Oncol. 2020, 10, 725.
- Choudhry, H.; Harris, A.L.; McIntyre, A. The tumour hypoxia induced non-coding transcriptome. Mol. Asp. Med. 2016, 47–48, 35–53.
- Ledoux, A.C.; Perkins, N.D. NF-kappaB and the cell cycle. Biochem. Soc. Trans. 2014, 42, 76–81.
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159.
- Scholz, C.C.; Cavadas, M.A.; Tambuwala, M.M.; Hams, E.; Rodriguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1beta-induced NF-kappaB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495.
- Cockman, M.E.; Lancaster, D.E.; Stolze, I.P.; Hewitson, K.S.; McDonough, M.A.; Coleman, M.L.; Coles, C.H.; Yu, X.; Hay, R.T.; Ley, S.C.; et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. USA 2006, 103, 14767–14772.
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-kappaB. Mol. Cell Biol. 2010, 30, 4901–4921.
- Melvin, A.; Mudie, S.; Rocha, S. Further insights into the mechanism of hypoxia-induced NFkappaB. [corrected]. Cell Cycle 2011, 10, 879–882.
- Bandarra, D.; Biddlestone, J.; Mudie, S.; Muller, H.A.; Rocha, S. Hypoxia activates IKK-NF-kappaB and the immune response in Drosophila melanogaster. Biosci. Rep. 2014, 34.
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell Biol. 1999, 19, 5785–5799.
- Joyce, D.; Albanese, C.; Steer, J.; Fu, M.; Bouzahzah, B.; Pestell, R.G. NF-kappaB and cell-cycle regulation: The cyclin connection. Cytokine Growth Factor Rev. 2001, 12, 73–90.
- Sheehy, A.M.; Schlissel, M.S. Overexpression of RelA causes G1 arrest and apoptosis in a pro-B cell line. J. Biol. Chem. 1999, 274, 8708–8716.
- Bash, J.; Zong, W.X.; Gelinas, C. c-Rel arrests the proliferation of HeLa cells and affects critical regulators of the G1/S-phase transition. Mol. Cell Biol. 1997, 17, 6526–6536.
- Yamada, K.; Ono, M.; Perkins, N.D.; Rocha, S.; Lamond, A.I. Identification and functional characterization of FMN2, a regulator of the cyclin-dependent kinase inhibitor p21. Mol. Cell 2013, 49, 922–933.
- Fitzpatrick, S.F.; Tambuwala, M.M.; Bruning, U.; Schaible, B.; Scholz, C.C.; Byrne, A.; O’Connor, A.; Gallagher, W.M.; Lenihan, C.R.; Garvey, J.F.; et al. An intact canonical NF-kappaB pathway is required for inflammatory gene expression in response to hypoxia. J. Immunol. 2011, 186, 1091–1096.
- Bandarra, D.; Biddlestone, J.; Mudie, S.; Muller, H.A.; Rocha, S. HIF-1alpha restricts NF-kappaB-dependent gene expression to control innate immunity signals. Dis. Model. Mech. 2015, 8, 169–181.
- Araki, K.; Kawauchi, K.; Tanaka, N. IKK/NF-kappaB signaling pathway inhibits cell-cycle progression by a novel Rb-independent suppression system for E2F transcription factors. Oncogene 2008, 27, 5696–5705.
- Ankers, J.M.; Awais, R.; Jones, N.A.; Boyd, J.; Ryan, S.; Adamson, A.D.; Harper, C.V.; Bridge, L.; Spiller, D.G.; Jackson, D.A.; et al. Dynamic NF-kappaB and E2F interactions control the priority and timing of inflammatory signalling and cell proliferation. Elife 2016, 5.

