 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Neil Grimsey | + 3704 word(s) | 3704 | 2021-04-21 07:36:09 | | | |
| 2 | Vivi Li | Meta information modification | 3704 | 2021-04-22 04:16:13 | | | | |
| 3 | Vivi Li | Meta information modification | 3704 | 2021-04-22 04:16:32 | | |
Video Upload Options
The mitogen-activated protein kinase (MAPK) p38 is an essential family of kinases, regulating responses to environmental stress and inflammation. There is an ever-increasing plethora of physiological and pathophysiological conditions attributed to p38 activity, ranging from cell division and embryonic development to the control of a multitude of diseases including retinal, cardiovascular, and neurodegenerative diseases, diabetes, and cancer. Despite the decades of intense investigation, a viable therapeutic approach to disrupt p38 signaling remains elusive. A growing body of evidence supports the pathological significance of an understudied atypical p38 signaling pathway. Atypical p38 signaling is driven by a direct interaction between the adaptor protein TAB1 and p38α, driving p38 autophosphorylation independent from the classical MKK3 and MKK6 pathways.
1. Introduction
The p38 mitogen-activated protein kinase (MAPK) family are critical cellular signaling regulators that drive many physiological and pathophysiological pathways. Therefore, it is not surprising that since their discovery in 1994 [1], over 45,000 research articles and reviews have been published describing the mechanism of p38 activation and the role of p38 during development and disease progression. The broader MAPK family includes c-Jun activated Kinase (JNK), extracellular signal-related kinase 1 and 2 (ERK1/2), and protein kinase B, also known as AKT kinase (AKT), all of which are critical in regulating a multitude of cellular processes from cell division to cell death and everything in between. Cellular stimuli/stress induces the activation of MAPKs, including hormones, growth factors, and cytokines, as well as environmental stressors such as osmotic shock, UV radiation, and ischemic injury [2]. As such, p38 MAPKs have been the subject of intense study to generate clinically effective therapeutics. Despite ongoing clinical trials for many diseases, including ischemic cardiac damage, COPD, multiple cancers, various neuropathies, and ARDS/COVID-19, only one non-selective p38 inhibitor (pirfenidone) has been approved for clinical use to treat idiopathic pulmonary fibrosis [3][4]. An underlying contributor to the loss of efficacy and on-target toxicity of these drugs is thought to be due to the ubiquitous and critical role p38 plays in normal physiology. Additionally, almost all current approaches have centered around therapeutics that target the ATP-binding site of p38 resulting in blockade of all p38 activity, both physiological and pathophysiological, regardless of the stimulus. Therefore, there is an increased focus on researching the downstream signaling targets of p38 induced only during disease progression, such as the critical inflammatory kinase MAPK-activated protein kinase 2 (MK2), or the alternative p38 activation pathways selectively induced during inflammation and disease progression (see Figure 1).
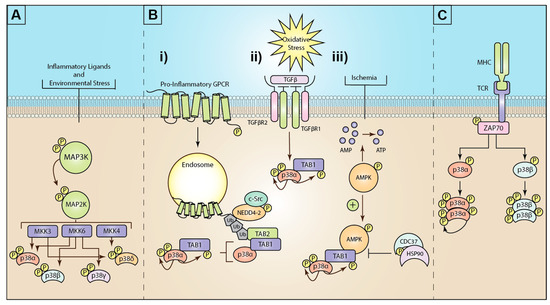
Figure 1. Mechanisms of mitogen-activated protein kinase (MAPK) p38 activation: (A) Inflammatory ligands and environmental stress trigger the activation of a three-tiered kinase cascade. Environmental or inflammatory ligands induce the activation of MAP3Ks through a complex array of different mechanisms. MAP3Ks then activate the critical MAP2Ks, MKK3, MKK6, or (less commonly) MKK4. These MAP2Ks can then differentially activate the four isoforms of p38 (α, β, γ, and δ). (B) The known mechanisms for atypical p38 signaling are (i) GPCR stimulation triggers G-protein dependent c-Src phospho-activation of the E3 ubiquitin ligase neural precursor cell expressed developmentally downregulated 4-2 (NEDD4-2). GPCRs recruit and are ubiquitinated by NEDD4-2. K63 ubiquitin chains recruit the ubiquitin-binding adaptor protein TAK1-binding protein 2 (TAB2). In turn, TAB2 then recruits TAB1, which binds and induces autophosphorylation of p38α. (ii) Oxidative stress triggers TGFβ activation, which drives TAB1 and p38 activation, although the exact mechanism is unclear. (iii) Ischemia or hypoxia events drive activation of AMP-activated protein kinase (AMPK), which in turn promotes the formation of the TAB1-p38α complex and p38α autophosphorylation. This process is negatively regulated by the heat shock protein 90 (HSP90)-Cdc37 complex. (C) T-cell receptor (TCR) ligation to major histocompatibility complex (MHC) drives intracellular activation of the src-family zeta-chain-associated protein kinase 70 (Zap70). Zap70 phosphorylates p38 at tyrosine 323, enabling autophosphorylation of p38α, or β.
In light of the sheer volume of p38 research articles and the wealth of excellent reviews available, it would be impractical and redundant to cover all aspects of p38 MAPK signaling. Therefore, this review will initially provide a brief overview of the history of p38 and the many roles it plays in disease progression. This will be followed by a more focused examination of the novel atypical p38 activation pathways, specifically including atypical p38 activation by GPCRs, their implications for disease progression, and therapeutic intervention. In comparison to classical p38 activity, atypical p38 signaling has been understudied with only 44 publications, however, this growing body of work represents a fresh perspective on p38 activity and function in disease.
2. Mechanisms of Atypical p38 Activation
MKK3/6 kinase activity is widely considered to be the primary mechanism for p38 phosphorylation. Nevertheless, there is a growing body of evidence to support alternative mechanisms for p38 activation (Figure 1). Two “atypical” or MKK3/6 independent mechanisms exist that facilitate activation of the p38α through autophosphorylation in cis, true autophosphorylation rather than phosphorylation of a neighboring p38 [5]. The first example of atypical p38 signaling was discovered in 2002, when p38α was shown to directly associate with transforming growth factor β-activated kinase 1 (TAK1) binding protein 1 (TAB1), an adaptor protein critical for both TGFβ and TAK1 signaling [6]. During osmotic stress responses [7], TAB1 is responsible for oligomerization and autophosphorylation of TAK1 after O-glycosylation, leading to TAK1 activation [8][9]. Conversely, in atypical p38 signaling, TAB1 binds directly and selectively to two discrete binding domains on p38α. Specifically, TAB1 residues 404–412 interact at a canonical site used by other p38 substrates, including MKK3 and MEF2a, and residues 389-394 bind to a non-canonical binding site on the c-terminal lobe of p38α. This site does not exist on any of the other p38 isoforms, and at the time of writing, no other proteins have been shown to bind to the same site on p38α [10][11]. The direct interaction of TAB1 with p38α induces a conformational change moving the active loop into the catalytic domain and enhancing ATP-binding, thus enabling cis-autophosphorylation of the active loop at Thr180 and Tyr182 [5]. Consequently, this leads to p38-induced phosphorylation of TAB1 at Ser423, downregulating TAB1 binding to TAK1 and inhibiting TAK1-mediated MKK3/6 activation [12]. Additional studies have also shown that TAB1 phosphorylation can alter its intracellular localization, where increased phosphorylation at S452/453/456/457 blocks its nuclear translocation causing TAB1 retention in the cytosol [13]. Intriguingly, TAB1 remains bound to p38α during atypical p38 activity, potentially suppressing the capacity of p38 nuclear translocation [11].
Reactive oxygen species are thought to be the initial driving force behind atypical p38 signaling in cardiac ischemia-reperfusion damage [11][5]. Similarly, cigarette smoke extract (CSE) induced oxidative stress in fetal tissue upregulating TGFβ production and resulting in TAB1-mediated p38 phosphorylation in a manner independent of TAK1 signaling or the ASK1-signalosome [14]. In a separate cardiac ischemia model, the TAB1-p38 interaction is upregulated in an AMPK-dependent manner [15] (Figure 1B ii). The interaction is negatively regulated by the HSP90/CDC37 chaperone complex in myocytes [16]. TAB1 expression is also negatively regulated by the E3 ligase itch through ubiquitin-mediated degradation. Where itch-deficient mice display dramatically increased dermal inflammation levels in an MKK3/6-independent manner [17]. The WW-domain in itch binds directly to a conserved PPXY motif in TAB1 (aa145–148). This interaction drives TAB1 K48-linked ubiquitination to regulate TAB1 turnover/degradation. TAB1 expression is significantly elevated in the absence of itch, leading to enhanced atypical p38 activation and increased cytokine production, including interleukin-6 (IL-6), interleukin-1beta (IL-1β), interleukin-11 (IL-11), and interleukin-19 (IL-19). Critically, Wang et al. in 2013 developed a peptide inhibitor fused to the HIV-TAT peptide, generating a cell-penetrating peptide inhibitor that selectively disrupts the TAB1 interaction with p38, substantially attenuating atypical p38 activation [18][19]. When used in the itch−/− mice, the peptide blocked atypical p38α signaling and dermal inflammation was significantly suppressed [17]. Further studies have shown that mutation of a critical proline proximal to the p38 binding peptide of TAB1 (P419) blocks TAB1 binding to p38α and prevents atypical p38α signaling [20][5][21], as does mutation of four key residues within the p38α-binding peptide of TAB1 (V390A, Y392A, V408G, and M409A) [11][5]. Critically, unlike the systemic knockout of TAB1 or p38α, which are embryonically lethal [22][23], the TAB1 knock-in (TAB1-KI) mouse displays no physiological abnormalities but is protected from myocardial ischemic damage [11].
This critical interaction provides a novel opportunity to further develop the peptide inhibitors or screen for small molecule inhibitors to target atypical p38 signaling selectively. Indeed, using a virtual small fragment screen, a group of functionalized adamantanes, specifically 3-amino-1-adamantanol, was found to bind to a critical hydrophobic pocket, forming hydrogen bonds with two key residues, leucine 222 and 234, in the non-canonical TAB1 binding site on p38α. Further screening found there to be three distinct fragment binding sites within the non-canonical binding site. Linking sulfonamide scaffolds to the adamantanol generated a small molecule with a high affinity to the three regions in the non-canonical binding site [10]. Additional development of these compounds will hopefully yield a viable therapeutic. However, it remains to be shown whether these lead hits can block atypical p38 signaling in cells or in vivo.
Despite these detailed studies describing the exact molecular mechanism of TAB1-p38α interaction and degradation, there are significant gaps in our understanding, specifically for how osmotic stress, oxidative stress, LPS, or inflammatory cytokines such as TNF-α and IL-1β initiate the TAB1-p38α interaction and atypical p38 signal transduction. Conversely, recent studies have shown that a family of G protein-coupled receptors (GPCRs) can initiate the TAB1-p38 interaction through a novel ubiquitin-driven pathway (described below and Figure 1B). This is the first example of a clearly defined mechanism for the induction of atypical p38 signaling and demonstrates conservation of the mechanism for at least four GPCRs critical for vascular inflammatory signaling and vascular homeostasis [20][24][25].
In addition to TAB1, a second discrete mechanism for p38 autophosphorylation has also been demonstrated through src-family zeta-chain-associated protein kinase 70 (Zap70). This pathway is critical for T-cell activation through a T-cell receptor (TCR) specific mechanism [26]. In contrast to TAB1-mediated autophosphorylation, p38α and p38β isoforms are phosphorylated at Tyr323 by ZAP70, leading to dimerization and mutual trans-autophosphorylation of the kinases at Thr180 alone. Tyr323 is located on the L16 loop of p38, facilitating this autophosphorylation by inducing a shift in the flexible phosphorylation lip of p38 (residues 171–183) [27]. Together, both TAB1- and ZAP70-mediated autophosphorylation of p38 reveal the kinase’s atypical activation in an MKK3/6-independent manner. The functional significance of these distinct activation mechanisms is still unclear. Additional studies are required to elucidate how atypical activation alters p38α substrate activation and induction of distinct signal transduction events. Notably, p38α is phosphorylated at the same sites in both classical MKK3/6-mediated and TAB1-mediated signaling, indicating that differential downstream signaling may instead be regulated in a spatiotemporal context rather than kinase functionality.
3. Activation of Atypical p38 by GPCRs
As the most extensive and versatile family of membrane proteins, G protein-coupled receptors (GPCRs) regulate many cellular pathways by activating MAPKs via G protein-dependent and -independent mechanisms [28][29][30][31][32]. Many of the GPCR families can activate p38α, but until recently, the mechanism for GPCR-mediated p38α activation remained unclear or was predicted to be controlled through the classical MKK3/6 pathway. However, several recent studies have linked vascular inflammatory GPCRs to the activation of the TAB1-dependent atypical p38 signaling pathway [20][24][6][25][33][34]. The initial studies examined thrombin-mediated activation of the protease-activated receptor 1 (PAR1) in vascular endothelial cells. The authors noted that after activation, PAR1 was ubiquitinated, despite being trafficked and degraded in a ubiquitin-independent manner [33][35][36][37]. α-Thrombin, activation of PAR1 induces the receptor to couple to the G protein subunits Gαq or Gα12/13 to induce activation of the proto-oncogene tyrosine-protein kinase c-Src (Src short for sarcoma) and subsequent activation of the E3 ubiquitin ligase, neural precursor cell expressed developmentally downregulated 4-2 (NEDD4-2) [24]. NEDD4-2 is one of a family of nine Homologous to E6-AP Carboxy Terminus (HECT) domain-containing E3 ligases and mediates the covalent coupling of ubiquitin to the intracellular c-tail or intracellular loops of GPCRs [20][25]. C-Src activates NEDD4-2 through tyrosine phosphorylation of a critical tyrosine residue, Y485, on a linker peptide between WW domain 2 and 3 (2,3 peptide). This 2, 3-linker peptide acts as a molecular switch that holds NEDD4-2 in an inactive conformation. Phosphorylation of Y485 by c-Src induces a conformational change that releases NEDD4-2 from an autoinhibited state. After activation, most likely at the plasma membrane, NEDD4-2 is recruited to PAR1, leading to PAR1 ubiquitination [24], although the exact mechanism as to how NEDD4-2 is recruited to PAR1 is unknown. Traditionally, GPCR ubiquitination serves as a sorting signal to cause endolysosomal trafficking and protein degradation [20][33]. However, in this case, NEDD4-2-mediated ubiquitination drives the recruitment of the TAB2-TAB1-p38 signaling complex [20][24][25][33]. TAB2 has an NP14 zinc finger (NZF) domain that binds to the lysine 63-linked NEDD4-2 ubiquitin chains and functions as an adaptor protein. It is predicted but has not been conclusively shown that TAB2 subsequently binds to and recruits TAB1 and p38α, inducing p38α autophosphorylation and TAB1 phosphorylation [20][38]. Interestingly, a structural homolog to TAB2, TAB3, is also able to bind to TAB1 to produce p38 pro-inflammatory signaling by GPCRs. However, it is not known what the contribution of each homolog is when expressed in the same cell or whether they are functionally redundant [25]. As stated above, the ubiquitinated endosomal receptors nucleate the formation and activation of the TAB1-p38α complex and increase TAB1 phosphorylation and stability [20]. It is still unclear whether GPCR-activated TAB1 sequesters p38 in the cytosol. Likewise, it is not known how TAB1-p38 signaling is terminated.
Importantly, this pathway is not unique just to PAR1 and α-thrombin. NEDD4-2 dependent regulation of atypical p38 signaling is also conserved for the purinergic receptor P2Y1. Furthermore, a recent study also demonstrated that the pathway is conserved for prostaglandin E2 (PGE2), histamine, ADP, and α-thrombin-mediated p38 activation and inflammatory cytokine production in primary human microvascular and macrovascular endothelial cells [25]. Additional studies are required to determine how many GPCRs utilize this pathway, whether atypical p38 signaling is critical for all cells, and how it selectively contributes to pathophysiological responses.
4. Pathophysiological Implications of Atypical p38 Signaling
Contrary to the highly studied MKK3/6-dependent pathway, the impact of TAB1-p38-dependent signaling in physiology and disease remains largely understudied with just 44 research articles on the subject (Table 1). As mentioned above, the recent development of the viable p38α-KI mouse [39] or the TAB1-KI mouse [11] suggests that perturbation of the atypical pathway is less critical for developmental and physiological signaling compared to the embryonically lethal systemic knockout of p38α or TAB1 [22][23]. It is perhaps then not surprising that atypical p38 activation has so far only been identified as a contributor to disease progression, which will be discussed below.
Table 1. Physiological roles of TAB1-dependent atypical p38 signaling.
| Disease | Mechanism of p38 Autophosphorylation |
Model | Specific Cell or Animal Line |
|---|---|---|---|
| Cardiovascular ischemia and reperfusion | TAB1-mediated | Murine in vivo [11][15][18][40][41] | MKK3−/− [15][41]; C57BL/6 [15][40]; Sprague Dawley [15]; Wistar [18]; TAB1 KI [11] |
| Murine in vitro [11][5][18][40][41][42] | H9c2 [41]; Sprague Dawley [18][40][42]; Wistar [18]; C57BL/6 [11][5] | ||
| Human in vitro [11][18][19][40][39] | HEK293 [11][18][40][39] | ||
| Structural modeling [10] | |||
| Myocardial infarction, amyloidosis, and cardiomyopathy | TAB1-mediated | Murine in vivo [43] | Sprague Dawley |
| Murine in vitro [16][43][44] | H9c2 [43]; Wistar [44] | ||
| Human in vitro [44] | Patient heart | ||
| Zebrafish in vivo [45] | |||
| General inflammation and cancer | TAB1-mediated | Murine in vivo [20][17][46] | BALB/c [46]; CD1/CD1 [20]; C57BL/6, Itch−/− [17] |
| Murine in vitro [20][17][46] | Vβ8.1, OT-II [46]; TAB1−/− [20]; C57BL/6, Itch−/− [17] | ||
| Human in vitro [20][24][25] | HUVEC [20][24][25]; HEK293 [20]; HDMEC [25] | ||
| Structural modeling [27][47] | |||
| Parasitic infection | TAB1-mediated | Murine in vivo [48] | BALB/c |
| Murine in vitro [48][49][50] | RAW264.9 [48]; MKK3−/− [49]; BALB/c [50] | ||
| Viral infection | TAB1-mediated | Murine in vitro [51] | C57BL/6, BC-1 |
| Human in vitro [52] | Huh7.5.1, HEK293, patient liver | ||
| Bacterial infection | TAB1-mediated | Human in vitro [53] | HPMEC |
| Shrimp [54] | |||
| Diabetes | TAB1-mediated | Murine in vitro [55][56] | β-TC6 [55][56]; Sprague Dawley, NMRI [56] |
| Human in vitro [56] | Islet | ||
| Leukocyte dysfunction | TAB1-mediated | Murine in vivo [57] | Vβ8.1 |
| Murine in vitro [57] | 2B4 | ||
| Human in vitro [58][59][60] | Patient blood | ||
| Pregnancy complications | TAB1-mediated | Murine in vitro [61] | CD-1 |
| Human in vitro [14][61] | Patient placenta | ||
| Other | TAB1-mediated | Murine in vitro [62][63] | MKK3−/−/6−/− [62]; MKK3−/− [63] |
| Human in vitro [6][13][21][64] | HEK293 [6][13][21][64]; MDA231 [64] | ||
| Structural modeling [65] | |||
| Immune system (T-Cell) modulation |
Zap70-mediated | Murine in vivo [26] | P116 |
| Murine in vitro [66][67][68][69] | Gadd45a−/− [66]; CD4SP [67]; C57BL/6 [68][69] | ||
| Human in vitro [26][70][71][72][73] | Jurkat, P116 | ||
| Chicken in vitro [72] | DT40 |
There is a growing awareness that atypical p38α activation plays a key role multiple p38 driven pathologies. The initial studies describing atypical p38α activation demonstrate its role in ischemic cardiac damage, ischemia-reperfusion injury, and amyloidosis. In an MKK3−/− ischemic mouse, the TAB1-p38 interaction was a leading contributor to necrosis in cardiomyocytes [41]. The role of atypical p38 was further confirmed in the progression of ischemic damage when a cell-penetrating inhibitor peptide was developed that reduced infarct size in ischemic rats [18]. Supporting this, the recent TAB1-KI mice where TAB1-induced autophosphorylation of p38 was genetically perturbed had significantly reduced infarction volume after induction of myocardial ischemia. Furthermore, the transphosphorylation of TAB1 was disabled [11], and cyclic GMP kinase 1 was found to inhibit TAB1-p38α to prevent apoptosis in cardiomyocytes during IR [40]. Additionally, basal activation of p38 autophosphorylation is suppressed by the HSP90/CDC37 complex where CD37 directly interacts with p38α [16]. Inhibition of HSP90 during cardiac stress is thought to dissociate HSP90 from p38α, enabling TAB1 interaction and p38α autophosphorylation to drive IL-6 and TNFα expression and cardiomyocyte apoptosis [16]. Additional studies have also shown that in a zebrafish model of amyloid light-chain (AL-LC) amyloidosis, AL-LC drives TAB1-p38α signaling causing cardiotoxic signaling, impaired cardiac function, pericardial edema, cell death, and subsequent heart failure [44][45].
Aside from the heart, p38 autophosphorylation has also been indicated in pathological inflammation in dermal disorders, preterm birth, and more broadly in vascular inflammation. In the itch−/− mice, TAB1 expression is significantly enhanced, leading to robust p38 autophosphorylation and subsequent increases in inflammatory cytokine expression, immune cell recruitment, and spontaneous skin lesions [17]. The use of the cell-penetrating peptide inhibitor significantly reduced these phenotypes, suggesting that itch-mediated p38 signaling could be exploited therapeutically [17]. In the field of reproductive biology, term and preterm parturition are tied to oxidative-stress and inflammatory TGF-β-induced TAB1-p38 activity resulting in amniochorion senescence [14]. Atypical p38 is also considered an essential component of the careful balance of endothelial mesenchymal transition (EndoMT) and mesenchymal endothelial transition (MEndoT) in human and murine amnion cells that contributes to the timing of parturition [61].
Vascular inflammation also directly activates GPCR-dependent p38 signaling in endothelial cells. In these studies, GPCR ligand α-thrombin induces endothelial barrier disruption driving vascular leakage and permeability. Additionally, recent studies of GPCR-mediated TAB1-p38 activity have demonstrated that it is conserved in multiple endothelial vascular beds and activated by a family of GPCR ligands associated with inflammation such as histamine, PGE2, ADP, and potentially many others [20][24][25]. While it has not yet been definitively shown, it stands to reason that any cell that expresses these GPCR receptors has the potential to induce atypical p38 signaling. This being the case, it will be essential to understand the role of GPCR signaling in fibroblasts, epithelial cells, mural/pericyte cells, and neuronal cells. Therefore, the impact of GPCR-induced atypical signaling is likely to play an, as of yet, undiscovered or overlooked role in many other vascular inflammatory diseases.
Beyond the vasculature, the role of atypical p38 is also explored in the modulation of the immune system by inflammatory ligands, attenuation of the TCR, and response to pathogens. Basophils and eosinophils isolated from healthy patients undergo p38 autophosphorylation in response to cytokine exposure from TNFα and GM-CSF, contributing to prolonged inflammation like that seen in pulmonary inflammatory disorders [58]. Conversely, TAB1-p38 interaction is also associated with maintaining anergic CD4+ T-cells through increased expression of TAB1 following antigen exposure and abrogating TCR [57]. Similarly, TAB1-p38 drives T-cell senescence via an AMPK-dependent regulatory pathway, resulting in downregulation of TCR signalosome [59]. AMPK also plays an essential role in the TAB1-p38 activation of HSP27 in simulated sepsis, maintaining vascular integrity [53]. Intracellular infection leading to TAB1-p38 activity was first shown in macrophages in mice infected with Toxoplasma gondii, resulting in pro-inflammatory IL-12 production specific to atypical signaling [49]. Leishmania infection results in parasite GP63-induced degradation of TAB1 to reduce p38 activation [50], the reversal of which sharply attenuates infection [48]. These studies suggest a vital role for the TAB1-p38 interaction in the host defense during intracellular pathogen infection.
Another example of atypical p38 activation comes from a recent study that demonstrated that multiple viruses utilize atypical p38 signaling to drive viral infections. Inhibition of TAB1-dependent p38 activation impaired hepatitis C virus (HCV) assembly and viral replication. This was also confirmed for severe fever with thrombocytopenia syndrome virus (SFTSV), herpes simplex virus type 1 (HSV-1), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [52]. Indeed, the p38 inhibitor losmapimod is currently in a clinical trial to treat SARS-CoV-2 (ClinicalTrials.gov ID: NCT04511819). It will be important for future studies to understand how atypical p38 signaling contributes to viral and bacterial infections and whether selective atypical p38 inhibitors could support current therapeutic regimens.
In the realm of type 1 diabetes, a link was found for TAB1-p38 interaction in the apoptosis of beta cells via oxidative stress by NO [55] and cytokine-induced beta-cell death [56]. These investigators noted that the effect of TAB1 signaling was specific to the TAB1α splicing product of the TAB1 gene located on chromosome 22, which has also been linked to systemic sclerosis and type 2 diabetes, hinting at a potential genetic component involving TAB1 mutation in the initiation of these diseases.
Contrary to TAB1-dependent signaling, Zap70-dependent activation of p38 is exclusive to T-cell activation via the TCR response, which is negatively regulated by p38 phosphorylation of upstream Zap70 [26][27]. However, a recent study also showed that TCR-mediated p38 activation occurs simultaneously through a classical kinase cascade and inflammatory augmentation by the alternative, atypical p38 activation. Intriguingly, it is suggested that uncoupling of the classical p38 activation mediated by the adaptor protein LAT and the guanine nuclear exchange factor, Son of Sevenless 1/2 (SOS1/2), reduced T-cell development and exacerbated autoimmune disease in mice [72]. At the same time, the genetic blockade of the TAB1-Zap70 suppressed T helper cell activation (TH1 and TH17) and expression of IFNγ and IL17. Indicating that both the classical and atypical p38 activation pathways could work synergistically to induce a balance between pro- and anti-inflammatory responses [72]. It is currently unclear whether there are some cases when TAB1-p38 activation may work in consort with MKK3/6, albeit in a TAK1-independent manner as TAB1 phosphorylation by p38 during atypical p38 signaling blocks TAB1′s interaction with TAK1 preventing TAB1-TAK1 dependent MKK3/6 activation [12].
References
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811.
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869.
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092.
- Valeyre, D.; Albera, C.; Bradford, W.Z.; Costabel, U.; King, T.E., Jr.; Leff, J.A.; Noble, P.W.; Sahn, S.A.; du Bois, R.M. Comprehensive assessment of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis. Respirology 2014, 19, 740–747.
- De Nicola, G.F.; Martin, E.D.; Chaikuad, A.; Bassi, R.; Clark, J.; Martino, L.; Verma, S.; Sicard, P.; Tata, R.; Atkinson, R.A.; et al. Mechanism and consequence of the autoactivation of p38α mitogen-activated protein kinase promoted by TAB1. Nat. Struct. Mol. Biol. 2013, 20, 1182–1190.
- Ge, B.; Gram, H.; Di Padova, F.; Huang, B.; New, L.; Ulevitch, R.J.; Luo, Y.; Han, J. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science 2002, 295, 1291–1294.
- Inagaki, M.; Omori, E.; Kim, J.Y.; Komatsu, Y.; Scott, G.; Ray, M.K.; Yamada, G.; Matsumoto, K.; Mishina, Y.; Ninomiya-Tsuji, J. TAK1-binding protein 1, TAB1, mediates osmotic stress-induced TAK1 activation but is dispensable for TAK1-mediated cytokine signaling. J. Biol. Chem. 2008, 283, 33080–33086.
- Scholz, R.; Sidler, C.L.; Thali, R.F.; Winssinger, N.; Cheung, P.C.; Neumann, D. Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process. J. Biol. Chem. 2010, 285, 25753–25766.
- Kishimoto, K.; Matsumoto, K.; Ninomiya-Tsuji, J. TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 2000, 275, 7359–7364.
- Nichols, C.; Ng, J.; Keshu, A.; Kelly, G.; Conte, M.R.; Marber, M.S.; Fraternali, F.; De Nicola, G.F. Mining the PDB for Tractable Cases Where X-ray Crystallography Combined with Fragment Screens Can Be Used to Systematically Design Protein-Protein Inhibitors: Two Test Cases Illustrated by IL1β-IL1R and p38α-TAB1 Complexes. J. Med. Chem. 2020, 63, 7559–7568.
- De Nicola, G.F.; Bassi, R.; Nichols, C.; Fernandez-Caggiano, M.; Golforoush, P.A.; Thapa, D.; Anderson, R.; Martin, E.D.; Verma, S.; Kleinjung, J.; et al. The TAB1-p38alpha complex aggravates myocardial injury and can be targeted by small molecules. JCI Insight 2018, 3.
- Cheung, P.C.; Campbell, D.G.; Nebreda, A.R.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J. 2003, 22, 5793–5805.
- Wolf, A.; Beuerlein, K.; Eckart, C.; Weiser, H.; Dickkopf, B.; Muller, H.; Sakurai, H.; Kracht, M. Identification and functional characterization of novel phosphorylation sites in TAK1-binding protein (TAB) 1. PLoS ONE 2011, 6, e29256.
- Richardson, L.; Dixon, C.L.; Aguilera-Aguirre, L.; Menon, R. Oxidative stress-induced TGF-beta/TAB1-mediated p38MAPK activation in human amnion epithelial cells. Biol. Reprod. 2018, 99, 1100–1112.
- Li, J.; Miller, E.J.; Ninomiya-Tsuji, J.; Russell, R.R., 3rd; Young, L.H. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ. Res. 2005, 97, 872–879.
- Ota, A.; Zhang, J.; Ping, P.; Han, J.; Wang, Y. Specific regulation of noncanonical p38alpha activation by Hsp90-Cdc37 chaperone complex in cardiomyocyte. Circ. Res. 2010, 106, 1404–1412.
- Theivanthiran, B.; Kathania, M.; Zeng, M.; Anguiano, E.; Basrur, V.; Vandergriff, T.; Pascual, V.; Wei, W.Z.; Massoumi, R.; Venuprasad, K. The E3 ubiquitin ligase Itch inhibits p38alpha signaling and skin inflammation through the ubiquitylation of Tab1. Sci. Signal. 2015, 8, ra22.
- Wang, Q.; Feng, J.; Wang, J.; Zhang, X.; Zhang, D.; Zhu, T.; Wang, W.; Wang, X.; Jin, J.; Cao, J.; et al. Disruption of TAB1/p38alpha interaction using a cell-permeable peptide limits myocardial ischemia/reperfusion injury. Mol. Ther. 2013, 21, 1668–1677.
- Pei, Y.J.; Wang, Q.Y.; Zhang, J.Y.; Guo, Y.H.; Feng, J.N. Characterization and Evaluation of Key Sites in the Peptide Inhibitor of TAB1/p38 alpha Interaction. Int. J. Pept. Res. Ther. 2018, 24, 225–233.
- Grimsey, N.J.; Aguilar, B.; Smith, T.H.; Le, P.; Soohoo, A.L.; Puthenveedu, M.A.; Nizet, V.; Trejo, J. Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J. Cell Biol. 2015, 210, 1117–1131.
- Zhou, H.; Zheng, M.; Chen, J.; Xie, C.; Kolatkar, A.R.; Zarubin, T.; Ye, Z.; Akella, R.; Lin, S.; Goldsmith, E.J.; et al. Determinants that control the specific interactions between TAB1 and p38alpha. Mol. Cell. Biol. 2006, 26, 3824–3834.
- Adams, R.H.; Porras, A.; Alonso, G.; Jones, M.; Vintersten, K.; Panelli, S.; Valladares, A.; Perez, L.; Klein, R.; Nebreda, A.R. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 2000, 6, 109–116.
- Komatsu, Y.; Shibuya, H.; Takeda, N.; Ninomiya-Tsuji, J.; Yasui, T.; Miyado, K.; Sekimoto, T.; Ueno, N.; Matsumoto, K.; Yamada, G. Targeted disruption of the Tab1 gene causes embryonic lethality and defects in cardiovascular and lung morphogenesis. Mech. Dev. 2002, 119, 239–249.
- Grimsey, N.J.; Narala, R.; Rada, C.C.; Mehta, S.; Stephens, B.S.; Kufareva, I.; Lapek, J.; Gonzalez, D.J.; Handel, T.M.; Zhang, J.; et al. A Tyrosine Switch on NEDD4-2 E3 Ligase Transmits GPCR Inflammatory Signaling. Cell Rep. 2018, 24, 3312–3323.e5.
- Grimsey, N.J.; Lin, Y.; Narala, R.; Rada, C.C.; Mejia-Pena, H.; Trejo, J. G protein-coupled receptors activate p38 MAPK via a non-canonical TAB1-TAB2 and TAB1-TAB3 dependent pathway in endothelial cells. J. Biol. Chem. 2019, 294.
- Salvador, J.M.; Mittelstadt, P.R.; Guszczynski, T.; Copeland, T.D.; Yamaguchi, H.; Appella, E.; Fornace, A.J., Jr.; Ashwell, J.D. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 2005, 6, 390–395.
- Diskin, R.; Lebendiker, M.; Engelberg, D.; Livnah, O. Structures of p38alpha active mutants reveal conformational changes in L16 loop that induce autophosphorylation and activation. J. Mol. Biol. 2007, 365, 66–76.
- Flower, D.R. Modelling G-protein-coupled receptors for drug design. Biochim. Biophys. Acta 1999, 1422, 207–234.
- Azzi, M.; Charest, P.G.; Angers, S.; Rousseau, G.; Kohout, T.; Bouvier, M.; Piñeyro, G. β-Arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 11406.
- Gutkind, J.S. Regulation of Mitogen-Activated Protein Kinase Signaling Networks by G Protein-Coupled Receptors. Sci. STKE 2000, 2000, re1.
- McDonald, P.H.; Chow, C.-W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.-T.; Davis, R.J.; Lefkowitz, R.J. β-Arrestin 2: A Receptor-Regulated MAPK Scaffold for the Activation of JNK3. Science 2000, 290, 1574.
- Shenoy, S.K.; Lefkowitz, R.J. Seven-transmembrane receptor signaling through beta-arrestin. Sci. STKE 2005, 2005, cm10.
- Burton, J.C.; Grimsey, N.J. Ubiquitination as a Key Regulator of Endosomal Signaling by GPCRs. Front. Cell Dev. Biol. 2019, 7, 43.
- Grimsey, N.J.; Trejo, J. Integration of endothelial protease-activated receptor-1 inflammatory signaling by ubiquitin. Curr. Opin. Hematol. 2016, 23, 274–279.
- Dores, M.R.; Chen, B.; Lin, H.; Soh, U.J.; Paing, M.M.; Montagne, W.A.; Meerloo, T.; Trejo, J. ALIX binds a YPX(3)L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J. Cell. Biol. 2012, 197, 407–419.
- Dores, M.R.; Grimsey, N.J.; Mendez, F.; Trejo, J. ALIX Regulates the Ubiquitin-Independent Lysosomal Sorting of the P2Y1 Purinergic Receptor via a YPX3L Motif. PLoS ONE 2016, 11, e0157587.
- Dores, M.R.; Lin, H.; Grimsey, N.J.; Mendez, F.; Trejo, J. The alpha-arrestin ARRDC3 mediates ALIX ubiquitination and G protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 2015, 26, 4660–4673.
- Kulathu, Y.; Akutsu, M.; Bremm, A.; Hofmann, K.; Komander, D. Two-sided ubiquitin binding explains specificity of the TAB2 NZF domain. Nat. Struct. Mol. Biol. 2009, 16, 1328–1330.
- Thapa, D.; Nichols, C.; Bassi, R.; Martin, E.D.; Verma, S.; Conte, M.R.; De Santis, V.; De Nicola, G.F.; Marber, M.S. TAB1-Induced Autoactivation of p38α Mitogen-Activated Protein Kinase Is Crucially Dependent on Threonine 185. Mol. Cell. Biol. 2018, 38.
- Fiedler, B.; Feil, R.; Hofmann, F.; Willenbockel, C.; Drexler, H.; Smolenski, A.; Lohmann, S.M.; Wollert, K.C. cGMP-dependent protein kinase type I inhibits TAB1-p38 mitogen-activated protein kinase apoptosis signaling in cardiac myocytes. J. Biol. Chem. 2006, 281, 32831–32840.
- Tanno, M.; Bassi, R.; Gorog, D.A.; Saurin, A.T.; Jiang, J.; Heads, R.J.; Martin, J.L.; Davis, R.J.; Flavell, R.A.; Marber, M.S. Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: Evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia. Circ. Res. 2003, 93, 254–261.
- Lu, G.; Kang, Y.J.; Han, J.; Herschman, H.R.; Stefani, E.; Wang, Y. TAB-1 modulates intracellular localization of p38 MAP kinase and downstream signaling. J. Biol. Chem. 2006, 281, 6087–6095.
- Du, C.S.; Yang, R.F.; Song, S.W.; Wang, Y.P.; Kang, J.H.; Zhang, R.; Su, D.F.; Xie, X. Magnesium Lithospermate B Protects Cardiomyocytes from Ischemic Injury Via Inhibition of TAB1-p38 Apoptosis Signaling. Front. Pharmacol. 2010, 1, 111.
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; Del Monte, F.; Ward, J.E.; Connors, L.H.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193.
- Mishra, S.; Guan, J.; Plovie, E.; Seldin, D.C.; Connors, L.H.; Merlini, G.; Falk, R.H.; MacRae, C.A.; Liao, R. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H95–H103.
- Ohkusu-Tsukada, K.; Toda, M.; Udono, H.; Kawakami, Y.; Takahashi, K. Targeted inhibition of IL-10-secreting CD25- Treg via p38 MAPK suppression in cancer immunotherapy. Eur. J. Immunol. 2010, 40, 1011–1021.
- Singh, R. Model Predicts That MKP1 and TAB1 Regulate p38α Nuclear Pulse and Its Basal Activity through Positive and Negative Feedback Loops in Response to IL-1. PLoS ONE 2016, 11, e0157572.
- Gupta, P.; Das, P.K.; Ukil, A. Antileishmanial effect of 18β-glycyrrhetinic acid is mediated by Toll-like receptor-dependent canonical and noncanonical p38 activation. Antimicrob. Agents Chemother. 2015, 59, 2531–2539.
- Kim, L.; Del Rio, L.; Butcher, B.A.; Mogensen, T.H.; Paludan, S.R.; Flavell, R.A.; Denkers, E.Y. p38 MAPK autophosphorylation drives macrophage IL-12 production during intracellular infection. J. Immunol. 2005, 174, 4178–4184.
- Hallé, M.; Gomez, M.A.; Stuible, M.; Shimizu, H.; McMaster, W.R.; Olivier, M.; Tremblay, M.L. The Leishmania surface protease GP63 cleaves multiple intracellular proteins and actively participates in p38 mitogen-activated protein kinase inactivation. J. Biol. Chem. 2009, 284, 6893–6908.
- Mikkelsen, S.S.; Jensen, S.B.; Chiliveru, S.; Melchjorsen, J.; Julkunen, I.; Gaestel, M.; Arthur, J.S.; Flavell, R.A.; Ghosh, S.; Paludan, S.R. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: Dependence on TRAF2 and TAK1. J. Biol. Chem. 2009, 284, 10774–10782.
- Cheng, Y.; Sun, F.; Wang, L.; Gao, M.; Xie, Y.; Sun, Y.; Liu, H.; Yuan, Y.; Yi, W.; Huang, Z.; et al. Virus-induced p38 MAPK activation facilitates viral infection. Theranostics 2020, 10, 12223–12240.
- Angé, M.; Castanares-Zapatero, D.; De Poortere, J.; Dufeys, C.; Courtoy, G.E.; Bouzin, C.; Quarck, R.; Bertrand, L.; Beauloye, C.; Horman, S. α1AMP-Activated Protein Kinase Protects against Lipopolysaccharide-Induced Endothelial Barrier Disruption via Junctional Reinforcement and Activation of the p38 MAPK/HSP27 Pathway. Int. J. Mol. Sci. 2020, 21, 5581.
- Wang, S.; Li, M.; Yin, B.; Li, H.; Xiao, B.; Lǚ, K.; Huang, Z.; Li, S.; He, J.; Li, C. Shrimp TAB1 interacts with TAK1 and p38 and activates the host innate immune response to bacterial infection. Mol. Immunol. 2017, 88, 10–19.
- Makeeva, N.; Roomans, G.M.; Welsh, N. Role of TAB1 in nitric oxide-induced p38 activation in insulin-producing cells. Int. J. Biol. Sci. 2006, 3, 71–76.
- Makeeva, N.; Roomans, G.M.; Myers, J.W.; Welsh, N. Transforming growth factor-beta-activated protein kinase 1-binding protein (TAB)-1alpha, but not TAB1beta, mediates cytokine-induced p38 mitogen-activated protein kinase phosphorylation and cell death in insulin-producing cells. Endocrinology 2008, 149, 302–309.
- Ohkusu-Tsukada, K.; Tominaga, N.; Udono, H.; Yui, K. Regulation of the maintenance of peripheral T-cell anergy by TAB1-mediated p38 alpha activation. Mol. Cell. Biol. 2004, 24, 6957–6966.
- Ten Hove, W.; Houben, L.A.; Raaijmakers, J.A.M.; Bracke, M.; Koenderman, L. Differential regulation of TNFα and GM-CSF induced activation of P38 MAPK in neutrophils and eosinophils. Mol. Immunol. 2007, 44, 2492–2496.
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972.
- Lanna, A.; Gomes, D.C.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.W.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363.
- Richardson, L.S.; Taylor, R.N.; Menon, R. Reversible EMT and MET mediate amnion remodeling during pregnancy and labor. Sci. Signal. 2020, 13.
- Kang, Y.J.; Seit-Nebi, A.; Davis, R.J.; Han, J. Multiple activation mechanisms of p38alpha mitogen-activated protein kinase. J. Biol. Chem. 2006, 281, 26225–26234.
- Kim, S.I.; Kwak, J.H.; Zachariah, M.; He, Y.; Wang, L.; Choi, M.E. TGF-β-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-β1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am. J. Physiol. Ren. Physiol. 2007, 292, F1471–F1478.
- Ge, B.; Xiong, X.; Jing, Q.; Mosley, J.L.; Filose, A.; Bian, D.; Huang, S.; Han, J. TAB1β (Transforming Growth Factor-β-activated Protein Kinase 1-binding Protein 1β), a Novel Splicing Variant of TAB1 That Interacts with p38α but Not TAK1. J. Biol. Chem. 2003, 278, 2286–2293.
- Xin, F.; Wu, J. Crystal structure of the p38α MAP kinase in complex with a docking peptide from TAB1. Sci. China Life Sci. 2013, 56, 653–660.
- Salvador, J.M.; Mittelstadt, P.R.; Belova, G.I.; Fornace, A.J.; Ashwell, J.D. The autoimmune suppressor Gadd45α inhibits the T cell alternative p38 activation pathway. Nat. Immunol. 2005, 6, 396–402.
- Dorn, T.; Kuhn, U.; Bungartz, G.; Stiller, S.; Bauer, M.; Ellwart, J.; Peters, T.; Scharffetter-Kochanek, K.; Semmrich, M.; Laschinger, M.; et al. RhoH is important for positive thymocyte selection and T-cell receptor signaling. Blood 2007, 109, 2346–2355.
- Round, J.L.; Humphries, L.A.; Tomassian, T.; Mittelstadt, P.; Zhang, M.; Miceli, M.C. Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF-kappaB transcription factors. Nat. Immunol. 2007, 8, 154–161.
- Liang, Y.; Yi, P.; Wang, X.; Zhang, B.; Jie, Z.; Soong, L.; Sun, J. Retinoic Acid Modulates Hyperactive T Cell Responses and Protects Vitamin A–Deficient Mice against Persistent Lymphocytic Choriomeningitis Virus Infection. J. Immunol. 2020, 204, 2984–2994.
- Hirata, S.; Fukamachi, T.; Sakano, H.; Tarora, A.; Saito, H.; Kobayashi, H. Extracellular acidic environments induce phosphorylation of ZAP-70 in Jurkat T cells. Immunol. Lett. 2008, 115, 105–109.
- Giardino Torchia, M.L.; Dutta, D.; Mittelstadt, P.R.; Guha, J.; Gaida, M.M.; Fish, K.; Barr, V.A.; Akpan, I.O.; Samelson, L.E.; Tagad, H.D.; et al. Intensity and duration of TCR signaling is limited by p38 phosphorylation of ZAP-70(T293) and destabilization of the signalosome. Proc. Natl. Acad. Sci. USA 2018, 115, 2174–2179.
- Jun, J.E.; Kulhanek, K.R.; Chen, H.; Chakraborty, A.; Roose, J.P. Alternative ZAP70-p38 signals prime a classical p38 pathway through LAT and SOS to support regulatory T cell differentiation. Sci. Signal. 2019, 12.
- Liu, J.; Guo, K.; Hu, L.; Luo, T.; Ma, Y.; Zhang, Y.; Lai, W.; Guo, Z. ZAP70 deficiency promotes reverse cholesterol transport through MAPK/ERK pathway in Jurkat cell. Mol. Immunol. 2019, 107, 21–28.


