 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dakota Goad | + 5594 word(s) | 5594 | 2021-04-21 07:44:37 | | | |
| 2 | Peter Tang | Meta information modification | 5594 | 2021-04-22 03:39:57 | | |
Video Upload Options
Pancreatic ductal adenocarcinoma (PDAC) is a devastating malignancy with poor prognosis and a dismal survival rate, expected to become the second leading cause of cancer-related deaths in the United States. Oncolytic virus (OV) is an anticancer approach that utilizes replication-competent viruses to preferentially infect and kill tumor cells. Vesicular stomatitis virus (VSV), one such OV, is already in several phase I clinical trials against different malignancies. VSV-based recombinant viruses are effective OVs against a majority of tested PDAC cell lines.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic neoplasm. It is a highly invasive malignancy, which forms a stromal desmoplastic reaction (desmoplasia), characterized by a dramatic increase in the proliferation of alpha-smooth muscle actin-positive fibroblasts and an increased production of many extracellular matrix components [1]. Family history, diabetes, and smoking are the most well-established risk factors for developing pancreatic cancer. Despite being only the 13th most common type of cancer, PDAC is the fourth-leading cause of cancer-related deaths and is predicted to become the second-leading cause of cancer-related death by 2030, as incidence increases while rates of survivorship remain stagnant due to late diagnosis and limited treatment options [2].
KRAS, CDKN2A, TP53, and SMAD4 serve as driver genes for PDAC development, and the vast majority of patients with fully established pancreatic cancer carry genetic defects in at least one of these genes [3]. Mutations in KRAS are present in 90% of PDAC tumors, 95% of PDAC tumors have mutations in CDKN2A (encodes p16), 50–75% in TP53, and SMAD4 (DPC4) is lost in approximately 50% of PDAC tumors [4]. Mutated KRAS oncogene leads to an abnormal, constitutively active, Ras protein. This results in aberrant activation of pathways responsible for survival and proliferation [5]. Inactivation of the tumor suppressor gene CDKN2A results in the loss of p16, a protein that serves as a regulator of the G1-S checkpoint of the cell cycle. Abnormalities in TP53 prevent it from acting as a tumor suppressor protein, including its important role as a regulator of DNA-damage checkpoints. Furthermore, many p53 mutants acquire devastating gain-of-function oncogenic activities, actually promoting cell survival, proliferation, invasion, migration, chemoresistance, and chronic inflammation. SMAD4 (DPC4) is related to the TGF-β signaling pathway, but some mutations result in abnormal signaling by TGF-β, a transforming growth factor receptor on the cell surface which can further increase the risk of cancer development by increasing the rate of cell growth and replication. In addition, germline mutations within BRCA2, BRCA1, ATM, and other genes were frequently identified in PDACs as inherited traits increasing susceptibility to PDAC development later in life [6][7]. These genes, especially when identified as being comorbid, are correlated with a significantly higher metastatic burden [8][9][10].
The primary treatments for PDAC include surgery, chemotherapy, radiotherapy, and palliative care [11]. Surgical resection still retains the greatest chance of success for potentially curing PDAC, however late-stage diagnosis due to ambiguous symptoms often results in tumors that are too-far progressed for surgery alone. Less than 25% of patients that present with PDAC are eligible for surgical resection, and 5-year survivorship of completely resected patients is approximately 37% [4]. In addition, even in patients where surgical resection was performed with either preparatory or subsequent adjuvant chemotherapy, there is a high rate of recurrence, and up to 80% of patients with recurrent PDAC will relapse with local and/or distant disease, which is associated with mortality within 2 years from diagnosis.
2. Major Challenges with Current PDAC Treatments
Since 1997, gemcitabine-based chemotherapy has been the standard first-line treatment for patients with unresectable locally advanced, or metastatic pancreatic cancer with a median survival rate of 4.4–5.6 months, especially when patients are not healthy enough for combination therapies [12]. Gemcitabine (dFdC) is an analog of deoxycytidine and a pro-drug that, once transported into the cell, must be phosphorylated by cellular deoxycytidine kinase to gemcitabine diphosphate (dFdCTP) and gemcitabine triphosphate (dFdCTP), both of which can inhibit processes required for DNA synthesis. Other commonly used chemotherapies for pancreatic cancer include 5-fluorouracil (5-FU), oxaliplatin, albumin-bound paclitaxel, capecitabine, cisplatin, irinotecan, and docetaxel [13][14]. Although several gemcitabine-based combination treatments exist, most have not considerably improved survival. While some combinatorial chemotherapy treatments, such as gemcitabine with erlotinib, have demonstrated potential for longer patient survival, the majority of patients eventually experience tumor progression due to the development of resistance, and therefore novel therapies are required, especially those that do not rely solely on chemotherapeutic drugs [15][16].
The mechanisms of de novo or inherent resistance of PDACs to chemo- or radiotherapeutics are not well understood. Several factors have been demonstrated to contribute to such resistance, including (i) multiple factors associated with the nature of the PDAC tumor microenvironment (TME) [17][18]; (ii) nucleoside transporters or/and nucleoside enzymes affecting drug uptake and metabolism [19]; (iii) hypoxia-inducible factor-1 alpha (HIF-1α) regulated glucose metabolism [20]; (iv) stromal-derived Insulin-like Growth Factors (IGFs) [21]; (v) abnormal expression of tumor-associated mucin proteins [22]; (vi) IFN-related DNA-damage resistance signature (IRDS) of some tumors [23]. The understanding of chemoresistance of PDACs to chemotherapy is very important, as at least some of these mechanisms could be also contributing to the resistance of PDACs to OV therapy.
The success of any treatment for PDAC is further complicated by the TME of PDAC, which is characterized by dense stroma comprised of abundant fibroblasts, hypoxia, and sparse vasculature. Moreover, the infiltration of tumor-promoting immune cells mediates immune evasion and promotes tumor progression. The stroma surrounding the tumor is primarily composed of pancreatic stellate cells (PSCs) which are activated by secreted factors such as TNFα, TGF-β, and interleukins 1, 2, 10, and themselves secrete mucins, collagen, fibronectin, and laminin in addition to some other factors, forming a thick extracellular matrix (ECM). This composition generates an incredibly dense physical barrier, to both host immune cells and potential therapeutics while also increasing interstitial pressure, which, when combined with sparse vasculature, forms a hypoxic environment, further inhibiting immune cells in terms of recruitment and effectiveness. PI3K/Akt, a key downstream mediator of many receptor tyrosine kinase signaling pathways involved in cell proliferation, migration, and inhibition of apoptosis, is phosphorylated under hypoxic conditions, along with MAPK (Erk), which regulates cell proliferation in response to various growth factors, which have been associated with resistance to gemcitabine [24][25]. The limits on antitumor immune cell recruitment also leads to T-cell exhaustion resulting in loss of cytotoxic effector function and further limits appropriate immune responses. SDF-1α/CXCR4 signaling-induced activation of the intracellular FAK-AKT and ERK1/2 signaling pathways and a subsequent IL-6 autocrine loop in cancer cells can further increase chemoresistance [26].
The low expression of nucleoside transporters (NT) and inactivity of nucleoside enzymes (NE) both affect the activity of gemcitabine. Low expression of a nucleoside transporter hENT1 restricts the uptake of gemcitabine, preventing its incorporation into the DNA of replicating cancer cells, and high expression of hENT1 is related to longer overall survival in pancreatic cancer patients [27][28]. The inactivation of deoxycytidine kinase (dCK), an enzyme responsible for the initial phosphorylation of gemcitabine, also mediates resistance. dCK is often inactivated in gemcitabine-resistant PDAC lines [29], and knockdown of dCK has been shown to lead to the development of resistance [30], while expression of a DCK transgene (along with uridine monophosphate kinase) sensitized pancreatic cancer cells to gemcitabine [31].
Pancreatic cancers metabolize glucose at higher rates and show higher expression of HIF-1α positively correlated with gemcitabine resistance [32][33]. HIF-1α increases glucose uptake and metabolism in the cell and is stabilized by MUC1, a common biomarker for cancers including PDAC [34]. Knockdown of HIF-1α in gemcitabine-resistant cells reduced tumor cell survival following gemcitabine treatment, and treatment with digoxin, and HIF-1α inhibitor, reduced glucose uptake and cell survival in cells treated with gemcitabine [35]. The increased glucose uptake under hypoxic conditions feeds into the glycolysis pathway and increases biomass; however, the exact mechanisms by which HIF-1α reduces sensitivity to chemotherapeutics have yet to be determined.
In addition, stromal-derived IGFs activate the insulin/IGF1R survival signaling pathway, reducing responsiveness to chemotherapeutics [36]. One proposed mechanism describes crosstalk between activated Insulin/IGF signaling pathways in PDAC. IGF-1 and IGF-1R, which are known to be abundantly expressed in the PDAC tissue, can stimulate β-cell proliferation and increase β-cell mass, increasing basal insulin production which may alter the trophic effects of the endocrine cells on the exocrine cells. Endocrine β-cells that express oncogenic K-ras can also be one potential progenitor for PDAC under chronic tissue inflammation [37]. This is further supported by evidence that demonstrates macrophages and myofibroblasts are the two major sources of IGFs within the pancreatic tumor microenvironment, and that chemoresistance is increased when cytotoxic agents increase M2-like macrophage infiltration [21]. For any novel therapies to be effective, they should be able to address most if not all of these challenges.
The structural composition of mucins produced by cells in certain cancers, such as breast and pancreatic cancers, has been suggested to limit immune cell recognition by blocking infiltration [38]. Similarly, the dense mucin mesh prevents cellular uptake of chemotherapeutics like gemcitabine and 5-FU within the tumor. MUC1 and MUV4 are overexpressed and aberrantly glycosylated in the majority of pancreatic tumors [39]. Kalra et al. demonstrated that the inhibition of mucin O-glycosylation enhanced the cytotoxic effects of 5-FU against human pancreatic cancer cell lines, but not against the mucin-deficient cell line [38]. They suggest that preventing the formation of the mucin facilitates the diffusion of drugs across the compromised mucus layer, improving intracellular drug uptake and enhancing cytotoxic drug action. Elevated MUC1 and MUC4 expression have also been correlated with greater degrees of resistance to gemcitabine [40]. It was also demonstrated that gemcitabine-resistant cells had accentuated the non-oxidative branch of the pentose phosphate pathway activity and increased pyrimidine biosynthesis, conferring resistance by increased dCTP production. MUC1 and MUC4 overexpression was also shown to upregulate mdr genes in pancreatic cancer cells, including ABCC1, ABCC3, ABCC5, and ABCB1 genes [39][41]. MUC4 expression was shown to be conversely correlated with the expression of hCNT1 and hCNT3 transporters, preventing uptake of chemotherapeutic drugs like gemcitabine, and hCNT1 is upregulated when MUC4 is inhibited, resulting in increased drug sensitivity [42]. Finally, MUC4-overexpressing CD18/HPAF-Src were not sensitive to gemcitabine, conferring resistance and survival advantages through erbB2-dependent and anti-apoptotic pathways [43]. Altogether, mucins including MUC1 and MUC4 have been demonstrated to be highly overexpressed and aberrantly glycosylated in pancreatic cancer cells, conferring resistance to various chemotherapies and the downregulation of these oncoproteins may represent a promising therapeutic strategy for reversing chemoresistance and reducing tumor progression and mass.
Type I IFN signaling is upregulated in some tumors responding to chemotherapy and can have antitumor as well as pro-tumor effects. The expression of a type I IFN-related DNA-damage resistance signature (IRDS) was reported to correlate with resistance to chemotherapy and radiotherapy in multiple cancer types. In breast cancer, the IRDS has been implicated in the development of chemoresistance, which may be another potential mechanism of resistance in PDACs as well [23]. The STAT1/IFN pathway transmits a cytotoxic signal either in response to DNA damage or to IFNs, such as in the case of viral infection. Cells with an IRDS (+) profile show constitutive activation of the STAT1/IFN pathway. Interestingly, this chronically activated state of the STAT1/IFN pathway may select against transmission of a cytotoxic signal, instead resulting in pro-survival signals mediated by STAT1 and other IRDS genes [23]. In agreement with this mechanism, STAT1 is highly upregulated in many cancers, including PDAC, and protects SCC-61 cells from ionizing radiation-mediated death [44]. STAT1 may also induce resistance with other DNA damage-based treatments, such as gemcitabine, and may transduce survival/growth signals that enhance tumor survival under some conditions [45]. Sensitivity to DNA damage is coupled with sensitivity to IFNs such that selection for resistance to one may lead to resistance to the other [46], which could prove to be a problem with not only chemo- and radiotherapies, but OV treatments as well.
3. Overview of Common Experimental Models to Study OV Therapy in PDAC
Oncolytic virus (OV) therapy is a relatively novel anticancer approach. Effective OV therapy is dependent on the oncoselectivity of OVs—their ability to preferentially infect, replicate in and kill infected cancer cells without damaging nonmalignant (“normal”) cells. The ideal OV therapy not only requires the direct lysis of cancer cells by the virus but also activates innate and adaptive anticancer immune responses [47] (Figure 1).
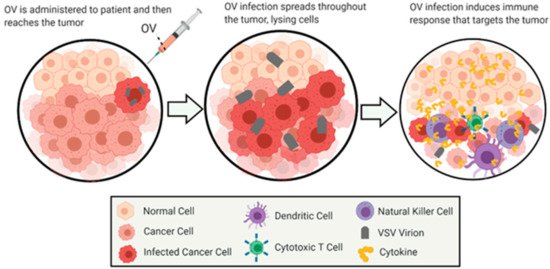
Figure 1. General Overview of Oncolytic Virotherapy. This figure demonstrates the general method of action for the treatment of cancer by oncolytic virotherapy using VSV as an oncolytic virus. The images depict the infection and oncolysis of malignant cells over time, followed by immunostimulation of cells invading the cleared area. The figure was created by authors with BioRender software (BioRender.com).
Preclinical PDAC models are critical for understanding the biology of PDAC, are platforms for developing novel strategies against PDAC, and are a necessary part of the drug development pipeline. There are several features of an ideal PDAC model system to develop clinically relevant OV therapy against PDAC: (1) the ability to test OV against different PDACs, characterized by various responsiveness to different therapies, including OV therapy; (2) the model should recapitulate a complex TME of PDACs; (3) tractability of the model, including the ability to trace both tumor cells and OV; (4) the ability to deliver OV systemically, as the PDAC are difficult to access; (5) the ability to detect and evaluate innate and adaptive immune responses against both tumor cells and OV. Unfortunately, there is no single PDAC model that successfully recapitulates all these critical features and challenges of the disease. However, there are numerous models for PDAC, each with unique advantages and disadvantages. Here, we will briefly review the advantages and disadvantages of various in vitro and in vivo models of PDAC and how they can contribute to the development of OV therapeutics.
4. Overview of the Current Progress in OV Therapy for PDAC
In 2015, the FDA approved Talimogene laherparepvec (T-VEC; Imlygic™), a genetically modified herpes simplex virus, to treat melanoma [48]. T-VEC is the first and still the only FDA-approved OV. However, numerous OVs are currently in preclinical studies and clinical trials for various malignancies, including PDAC. Table 1 and Table 2 summarize the results of preclinical studies (Table 1) and clinical trials (Table 2) of various oncolytic viruses against pancreatic cancer.
Table 1. Preclinical studies of oncolytic viruses against pancreatic cancer.
|
Oncolytic Virus Backbone |
Oncolytic Virus Name |
Brief Description of the Results of the Study |
|---|---|---|
|
Herpes virus |
HSV-GFP (expressing NeoR and EGFP) |
The pancreatic cancer lines MIAPACA and PANC-1 exhibited definite cytopathic effects upon infection in vitro (hPDAC cell lines) [49]. |
|
HSV2 L1BR1 (US3-deficient) |
US3-deficient HSV virus L1BR1 demonstrates a favorable characteristic regarding the induction of apoptosis in vitro (hPDAC cell lines) [50]. |
|
|
HSV-1 (DF3gamma134.5) |
The DF3/MUC1 promoter is shown to enhance the oncolytic activity of HSV-1 mutants in vitro (hPDAC cell lines) [51]. |
|
|
R3616: γ134.5 hrR3: UL39 |
In vivo evaluation of two herpes virus mutants in combination with gemcitabine show complex interactions that can benefit or inhibit oncolytic activity (hPDAC IP into BALB/C nude, immunodeficient) [52]. |
|
|
FusOn-H2 |
FusOn-H2 has potent activity against human pancreatic cancer xenografts in vivo (hPDAC SC or OT into Hsd nude, immunodeficient) [53]. |
|
|
NV1066 |
5-FU and gemcitabine determined in vivo to potentiate oncolytic herpes viral replication and cytotoxicity across a range of clinically achievable doses in the treatment of human pancreatic cancer (hPDAC cell lines) [54]. |
|
|
HSV(GM-CSF) |
Injection of the recombinant mouse HSV encoding GM-CSF resulted in a significant reduction in tumor growth (mPDAC cells SC into C57BL6, immunocompetent) [55]. |
|
|
Ad-DHscIL12 |
Expression of IL-12 in the context of a hypoxia-inducible oncolytic adenovirus is effective against pancreatic cancer in vivo (hamPDAC cells SC into nude, immunodeficient) [56]. |
|
|
NV1020 G2O7 |
NV1020 and G2O7 effectively infect and kill human pancreatic cancer cells in vitro and in vitro (hPDAC cell lines) [57]. |
|
|
Adenovirus |
AdΔCAR-WT, AdΔCAR-SYE, AdΔCAR-IVR |
An in vitro tumor-targeting strategy using an adenovirus library for optimization of oncolytic adenovirus therapy (hPDAC cells SC into BALB/C nude, immunodeficient) [58]. |
|
VCN-01 |
Oncolytic adenovirus VCN-01 shows an efficacy-toxicity profile in vivo (hPDAC cells SC into BALB/C nude, immunodeficient) [59]. |
|
|
AdDeltaE1B19K dl337 |
Novel adenoviral mutants demonstrate the ability to improve efficacy of DNA-damaging drugs such as gemcitabine in vitro and in vivo (hPDAC cells SC into ICRF nude, immunodeficient) [60]. |
|
|
WtΔE3ADP-Luc, WtΔE3ADP-IFN |
Adenoviral death protein and fiber modifications significantly improved oncolysis in vitro and in vivo (hPDAC cells SC into NCr nude, immunodeficient) [61]. |
|
|
Ad5PTDf35 |
This vector shows dramatically increased transduction capacity of primary human cell cultures including T cells, monocytes, macrophages, dendritic cells, pancreatic islets and exocrine cells, mesenchymal stem cells and tumor initiating cells (hPancreatic islet cells) [62]. |
|
|
ONYX-411 |
These findings indicate that Internavec can generate a two-pronged attack on tumor cells through oncogene knockdown and viral oncolysis, resulting in a significantly enhanced antitumor outcome in vitro and in vivo (hPDAC cells SC into nude, immunodeficient) [63]. |
|
|
ZD55-lipocalin-2 |
ZD55-lipocalin-2 may serve as a potent anticancer drug for pancreatic cancer therapy, especially for patients who have pancreatic adenocarcinoma with KRAS mutations as demonstrated in vitro (hPDAC cell lines) [64]. |
|
|
LOAd703 |
LOAd703 is a potent immune activator that modulates the stroma to support antitumor responses in vitro and in vivo (hPDAC cells SC into C57BL/6 nude, immunodeficient) [65]. |
|
|
OAd-hamIFN |
Combination treatment of chemoradiation with IFN-expressing OAd demonstrates enhanced cancer-killing efficacy in vitro and in vivo (hamPDAC cells SC into Golden Syrian hamsters, immunocompetent) [66]. |
|
|
OAd-TNFa-IL2 |
Ad-mTNFa-mIL2 increased immune cell infiltration to the tumor and altered host tumor immune status in vivo (hPDAC cells SC into NSG, immunodef and mPDAC cells SC into C57BL/6, immunocompetent) [67]. |
|
|
YDC002 |
YDC002 combined with gemcitabine significantly attenuated the expression of major ECM components including collagens, fibronectin, and elastin in tumor spheroids and xenograft tumors compared with gemcitabine alone, resulting in potent induction of apoptosis, gemcitabine-mediated cytotoxicity, and an oncolytic effect through degradation of tumor ECM in vivo (hPDAC cells SC into BALB/C nude, immunodeficient) [68]. |
|
|
AdΔΔ |
AdΔΔ has low toxicity to normal cells while potently sensitizing pancreatic cancer cells to DNA-damaging drugs in vivo (hPDAC cells SC into C57BL/6 nude, immunodeficient) [69]. |
|
|
dl922-947 |
dl922-947 is effectively able to elicit an anti-tumoral response in vivo when combined with 5-FU or gemcitabine (hPDAC cells SC into C57BL/6 nude, immunodeficient) [70]. |
|
|
Delta-24-RGD |
Delta-24-RGD significantly inhibited tumor growth in combination with phosphatidylserine targeting antibody in vivo (hPDAC cells SC into nude, immunodeficient) [71]. |
|
|
Ad5-3Δ-A20T |
Ad5-3Δ-A20T is highly selective for αvβ6 integrin-expressing pancreatic cancer cells for improved targeting of pancreatic cancer in vitro (hPDAC cell lines, 3D culture) [72]. |
|
|
ICOVIR15 |
Arming the oncolytic adenovirus ICOVIR15 with miR-99b or miR-485 enhances its fitness and its antitumoral activity in vitro (human lung, breast, colorectal, prostate cancer cell lines) [73] |
|
|
Ad5-yCD/mutTK(SR39)rep-ADP |
Ad5-yCD/mutTK(SR39)rep-ADP improves oncolysis in vitro and in vivo in combination with radiotherapy (hPDAC cells IM into CD-1 athymic, immunodeficient) [74]. |
|
|
Myxoma Virus |
vMyxgfp |
vMyxgfp had the ability to infect all pancreatic cancer cell lines tested in vitro (hPDAC cell lines) [75]. |
|
Reovirus |
Reolysin |
Reolysin treatment stimulated selective reovirus replication and decreased cell viability in KRas-transformed immortalized human pancreatic duct epithelial cells and pancreatic cancer cell lines in vitro and in vivo (hPDAC cells SC into nude, immunodeficient) [76]. |
|
Vaccinia Virus |
oVV-Smac |
oVV-Smac is indicated to have a synergistic effect in combinatorial treatment with gemcitabine in vitro and in vivo (hPDAC cells SC into BALB/C nude, immunodeficient) [77]. |
|
VV-HBD2-lacZ |
These results indicate that HBD2-expressing VV recruited plasmacytoid DCs (pDCs) to the tumor location, leading to cytotoxic T cell response against the tumor, and thus inhibited tumor growth in vitro and in vivo (murine melanoma cells SC into C57BL/6, immunocompetent) [78]. |
|
|
GLV-1h68 |
GLV-1h68 was able to infect, replicate in, and lyse tumor cells in vitro and in vivo (hPDAC cells SC into BALB/C nude, immunodeficient) [79]. |
|
|
VVLΔTK-IL-10 |
VV expressing IL-10 demonstrates enhanced anti-tumor efficacy in vivo (GEMM (KPC), immunocompetent) [80]. |
|
|
VVhEA |
The novel Lister strain of vaccinia virus armed with the endostatin-angiostatin fusion gene displayed inherently high selectivity for cancer cells, sparing normal cells both in vitro and in vivo, with effective infection of tumors (hPDAC cells SC into BALB/C nude, immunodeficient) [81]. |
|
|
Parvovirus |
H-1PV |
H-1PV in combination with gemcitabine enhanced anti-tumor activity of NK cells and effects included reduction in tumor growth, prolonged survival of the animals, and absence of metastases on CT-scans in vitro (hPDAC cell lines) [82]. |
|
H-1PV |
In ex vivo human models, H-1PV reinforced drug-induced tumor cell killing and effective immunostimulation (human melanoma cell lines) [83]. |
|
|
H-1PV |
The combination treatment of H-1PV and histone deacetylase inhibitors (HDACIs) such as valproic acid (VPA)acts synergistically to kill a range of human cervical carcinoma and pancreatic carcinoma cell lines by inducing oxidative stress, DNA damage and apoptosis in vitro and in vivo (hPDAC cells SC into NOD/SCID nude, immunodeficient) [84]. |
|
|
Measles Virus |
MV-PNP-anti-PSCA |
PNP, which activates the prodrug fludarabine effectively, enhanced the oncolytic efficacy of the virus on infected and bystander cells in vitro and in vivo (hPDAC cells SC into NOD/SCID nude, immunodeficient) [85]. |
|
MeV |
The chemovirotherapeutic combination of gemcitabine plus oncolytic MeV resulted in improved tumor reduction in vitro (hPDAC cell lines) [86]. |
|
|
Newcastle Disease Virus |
NDV |
NDV infection was successful in all evaluated PA cell lines in vitro, however the resultant replication kinetics and cytotoxic effects differed (hPDAC cell lines) [87]. |
|
NDV |
Infection with NDV activated immune cells which successfully elicited an anti-tumor response in vitro. However, activated NK cells that are abundant in Panc02 tumors lead to outgrowth of nonimmunogenic tumor cells with inhibitory properties (hPDAC cells OT into C57BL/6, immunocompetent) [88]. |
|
|
MTH-68/H |
MTH-68/H selectively kills tumor cell cultures in vitro by inducing endoplasmic reticulum stress leading to p53-independent apoptotic cell death (hPDAC cell lines) [89]. |
|
|
Poxvirus |
CF33 |
CF33 caused rapid killing of six pancreatic cancer cells lines in vitro, releasing damage-associated molecular patterns, and regression of tumors in vivo (human colorectal cells SC into Hsd nude, immunodeficient) [90]. |
|
Influenza Virus |
PR8, H5N1, H7N3, H4N8, H7N7, H5N1 HP, H7N1 HP |
IAV significantly inhibited tumor growth following intratumoral injection without inducing apoptosis in nonmalignant cells in vivo (hPDAC cells SC into SCID, immunodeficient) [91]. |
|
Rhabdovirus |
M51R-VSV |
M51R-VSV treatment appears to induce antitumor cellular immunity in vivo (hPDAC cells SC into C57BL/6 nude, immunodeficient) [92]. |
|
VSV-FH |
VSV-FH can induce potent oncolysis in hepatocellular and pancreatic cancer cell lines in vivo (hPDAC cells SC into athymic nude, immunodeficient) [93]. |
|
|
VSV-ΔM51 |
VSV showed oncolytic abilities superior to those of other viruses, and some cell lines that exhibited resistance to other viruses were successfully killed by VSV in vitro (hPDAC cell lines) [94]. |
|
|
VSV-mp53, VSV-ΔM-mp53 |
VSV expressing p53 exhibited enhanced oncolytic action, while VSV-ΔM-mp53 was extremely attenuated in vivo due to p53 activating innate immune genes (hPDAC cells IV into BALB/C nude, immunodeficient) [95]. |
|
|
VSV-ΔM51 |
VSV recombinants induced robust apoptosis in cells with defective IFN signaling, however cell lines constitutively expressing high levels of IFN-stimulated genes (ISGs) were resistant to apoptosis even when VSV replication levels were dramatically increased by Jak inhibitor I treatment in vitro (hPDAC cell lines) [96]. |
|
|
VSV-WT, VSV-rM51R-M |
Recombinant M51R-M (rM51R-M) virus induces apoptosis much more rapidly in L929 cells than viruses expressing WT M protein by a distinct method in vitro (murine fibroblast cell lines) [97]. |
|
|
VSV-ΔM51 |
TPCA-1 (IKK-β inhibitor) and ruxolitinib (JAK1/2 inhibitor), as strong enhancers of VSV-ΔM51 replication and virus-mediated oncolysis in all VSV-resistant cell lines in vitro (hPDAC cell lines) [98]. |
|
|
VSV |
Combining VSV with ruxolitinib and Polybrene or DEAE-dextran successfully broke the resistance of HPAF-II cells to VSV by simultaneously improving VSV attachment and replication in vitro (hPDAC cell lines) [99]. |
|
|
VSV, VSV-GFP, VSV-ΔM51-GFP |
In vivo administration of VSV-ΔM51-GFP resulted in significant reduction in tumor growth for tested mouse PDA xenografts and antitumor efficacy was further improved when the virus was combined with gemcitabine (mPDAC cells SC into C57BL/6, immunocompetent) [100]. |
|
|
VSV-p53wt, VSV-p53-CC |
Two independently evolved VSVs obtained identical glycoprotein mutations, K174E and E238K; these acquired G mutations improved VSV replication, at least in part due to improved virus attachment to SUIT-2 cells, as determined in vitro (hPDAC cell lines) [101]. |
hPDAC = human PDAC, mPDAC = mouse, SC = subcutaneous injection; OT = orthotopic injection, IP = intraperitoneal injection; IV = intravenous injection.
Table 2. Clinical trials featuring oncolytic viruses against pancreatic cancer.
|
Oncolytic Virus Backbone |
Oncolytic Virus Name |
Brief Description of the Clinical Trial |
|---|---|---|
|
Adenovirus |
ONYX-015 (dl1520) |
ONYX-015 injection via EUS into pancreatic carcinomas by the transgastric route with prophylactic antibiotics is feasible and generally well tolerated either alone or in combination with gemcitabine [102]. |
|
ONYX-015 (dl1520) |
Intratumoral injection of an E1B-55 kDa region-deleted adenovirus into primary pancreatic tumors was feasible and well-tolerated at doses up to 10(11) PFU (2 x 10(12) particles), but viral replication was not detectable [103]. |
|
|
Ad5-yCD/mutTKSR39rep-ADP |
A combination of intratumoral Ad5-DS and gemcitabine is safe and well tolerated in patients with LAPC [104]. |
|
|
Ad5-yCD/mutTKSR39rep-hIL12 |
Ongoing clinical trial, no results posted to date. NCT03281382. |
|
|
LOAd703 |
Ongoing clinical trial, no results posted to date. NCT02705196. |
|
|
VCN-01 |
Ongoing clinical trial, no results posted to date. NCT02045589. |
|
|
VCN-01 |
Ongoing clinical trial, no results posted to date. NCT02045602. |
|
|
Herpesvirus |
T-VEC |
EUS-guided FNI of T-VEC in advanced pancreatic ca, at initial doses of 104 to 106 PFU/mL followed by up to 107 PFU/mL, was feasible and tolerable. Evidence of biologic activity was observed [105]. |
|
T-VEC |
Ongoing clinical trial, no results posted to date. NCT03086642. |
|
|
HF10 |
HF10 direct injection under EUS-guidance in combination with erlotinib and gemcitabine was a safe treatment for locally advanced pancreatic cancer [106]. |
|
|
HF10 |
Ongoing clinical trial, no results posted to date. NCT03252808. |
|
|
OrienX010 |
Ongoing clinical trial, no results posted to date. NCT01935453. |
|
|
Reovirus |
Reolysin |
Pelareorep was safe but ineffective when administered with carboplatin/paclitaxel, regardless of KRAS mutational status. Immunologic studies suggest that chemotherapy backbone improves immune reconstitution and that targeting remaining immunosuppressive mediators may improve oncolytic virotherapy [107]. |
|
Reolysin |
PD analysis revealed reovirus replication within pancreatic tumor and associated apoptosis. Upregulation of immune checkpoint marker PD-L1 suggests future consideration of combining oncolytic virus therapy with anti-PD-L1 inhibitors [108]. |
|
|
Reolysin |
Pelareorep and pembrolizumab added to chemotherapy did not add significant toxicity and showed encouraging efficacy [109]. |
|
|
Reolysin |
Ongoing clinical trial, no results posted to date. NCT01280058. |
|
|
Parvovirus |
ParvOryx |
The drug was safe and well-tolerated and showed a promising profile of anti-tumor effects and signs of clinical efficacy, i.e., prolonged survival. However, the optimum dose as well as the most appropriate route and schedule of administration have to be further investigated [110]. |
PFU = plaque-forming unit.
Preclinical studies demonstrated that a wide range of different viruses could be efficient OVs against PDAC (Table 1).
Additionally, gene therapy targets, such as oncogene knockdown, insertion of functional tumor-suppressor genes, and expression of functional RNAs also demonstrate improved cancer-killing efficacy when combined with OV. One method uses adenoviruses and adeno-associated viruses to deliver apoptotic genes to tumor cells. Such gene therapy using Adenovirus subtype 5 mediates rat insulin promoter directed thymidine kinase (A-5-RIP-TK)/ganciclovir (GCV) gene therapy resulting in significantly enhanced cytotoxicity to both Panc1 and MiaPaCa2 pancreatic cancer cells in vitro [111]. Another review explored the potential use of OV expressing functional p53 [112]. Another method would use OV to deliver siRNA transgenes for oncogenetic knockdown, such as ONYX-411-siRNAras expressing a mutant K-ras siRNA which significantly reduced K-ras mRNA expression at 48 h posttreatment and improved oncolytic activity [63]. The inclusion of an endostatin-angiostatin fusion gene in VVhEA also showed significant antitumor potency in vivo [81].
There have been many experiments screening for more effective virotherapies within available libraries, and modulated viruses such as the adenovirus AdΔCAR-SYE has been shown to significantly suppress tumor growth, and complete regression of tumors was observed in vivo [58]. In addition, more efficient and tumor-specific targeting peptides and OV could be identified by using additional libraries, and modifications to existing OV based on these findings are also promising. Such modifications have been shown to be effective, with the adenoviruses VCN-01 variants ICOVIR-15K and ICOVIR-17 [59]. As discussed previously, the ability of the OV to modulate the ECM was observed, as tumors treated with VCN-01 showed a dramatic decrease in the intratumoral HA content [59]. Other adenoviral variants such as Ad5PTDf35(pp65) have also demonstrated T-cell stimulation and dendritic cell (DC) modulation to increase efficient transduction within a human context [62].
Combinatorial treatments of chemotherapies, or chemovirotherapy, such as OV paired gemcitabine, have demonstrated improved oncolytic capabilities in vitro and in vivo than either treatment on their own. In vitro and in vivo studies showed that myxoma virus (MYXV) and gemcitabine therapies can be combined sequentially to improve the overall survival in intraperitoneal dissemination (IPD) models of pancreatic cancer [113]. The addition of chemotherapies to OV therapy using a combination of an oncolytic herpes simplex virus-1 mutant NV1066 with 5-FU increased viral replication up to 19-fold compared with cells treated with virus alone, and similar results were achieved by the addition of gemcitabine [54]. Similarly, oVV-Smac combined with gemcitabine greater cytotoxicity and potentiated apoptosis [77]. H-1PV combined with cisplatin, vincristine or sunitinib induced effective immunostimulation via a pronounced DC maturation, better cytokine release and cytotoxic T-cell activation [83]. The addition of gene targets alongside chemovirotherapy has also shown greater cytotoxic efficiency, as with VV-ING4 in combination with gemcitabine [79]. Even in cell lines that demonstrate resistance to viral infection, resistance can be broken with simultaneous treatments. Viruses like VSV rely on nonspecific interactions with the cell surface during the earliest stages of infections, and polycations have been shown to improve viral production and increase oncolysis by increasing the amount of virus interacting with cells during attachment [99]. Additionally, the use of JAK inhibitors like ruxotinilib and IKK inhibitors like TPCA-1 have also been shown to increase viral reproduction and oncolysis [98][99]. Other potential combinatorial treatment regimens could include radiovirotherapy or chemoradiotherapy.
Some of these treatment methodologies are already being tested in clinical trials. Table 2 describes the OV currently being tested in clinical trials, and while some are still underway, OV including ONYX-15, AD5-yCD, and T-VEC are well-tolerated, and in some cases, biologically active, either alone or in combination chemovirotherapies [102][103][104][105][106][109].
5. Understanding Molecular Mechanisms of Responsiveness and Resistance of PDACs to VSV-Based OV Therapy
VSV is a prototypic nonsegmented negative-strand (NNS) RNA virus (order Mononegavirales, family Rhabdoviridae). VSV is a promising oncolytic virus against various malignancies, and it has several advantages as an OV [114][115][116]: (i) its basic biology and interaction with the host have been extensively studied. The oncoselectivity of VSV is mainly based on VSV’s high sensitivity to Type I interferon (IFN) mediated antiviral responses (and therefore inability to replicate in healthy cells), while it can specifically infect and kill tumor response cells, most of which lack effective Type I IFN responses; (ii) although WT VSV can cause neurotoxicity in mice, nonhuman primates, several VSV recombinants, including VSV-∆M51, have been generated which are not neurotropic but retain their OV activity; (iii) VSV has a broad tropism for different types of cancer cells (including PDACs), as its primary mode of entry into a host cell utilizes binding of the VSV-G protein to LDLR, which is ubiquitous, and VSV-G is also capable of using other common surface molecules for cell entry [115]; (iv) there is no preexisting immunity against VSV in most humans; (v) replication occurs in the cytoplasm without risk of host cell transformation; (vi) cellular uptake occurs rapidly; (vii) VSV has a small, easily manipulated genome, and novel VSV-based recombinant viruses can be easily engineered via reverse genetics to improve oncoselectivity, safety, oncotoxicity, and to work synergistically with host immunity and/or other therapies in a specific tumor environment (e.g., PDAC); (viii) as other members of the order Mononegavirales, and compared to positive-strand RNA viruses, VSV is less likely to mutate, and our recent study demonstrated long-term genetic stability of VSV recombinants carrying large transgenes [101]. All these and other advantages make VSV a promising candidate OV for PDAC treatment, and we have shown that VSV is effective against the majority of PDAC cell lines in vitro and in vivo [117][94]. Importantly, several phase I clinical trials using VSV against different malignancies are in progress (ClinicalTrials.gov for trials NCT03647163, NCT02923466, NCT03120624, NCT03865212, and NCT03017820).
VSV exhibits inherent oncotropism based largely on defective or reduced type I IFN responses, as specific genes associated with type I IFN responses are downregulated or functionally inactive [95][118]. In addition, IFN signaling can be inhibited by MEK/ERK signaling or by epigenetic silencing of IFN-responsive transcription factors IRF7 or IRF5 [119][120]. However, some PDACs do not have these defects and resist VSV infection like normal cells, which are sensitive to IFN-α treatment and capable of secreting type I IFNs following VSV infection [121].
There has been a demonstration of neurotoxicity in mice infected intranasally or intracranially, demonstrating a need for methods of improvement of VSV oncoselectivity and neurotropic safety without compromising oncolytic ability. There are at least eight approaches demonstrated to address these needs [114][115][116]: (i) mutating the VSV M protein; (ii) VSV-directed IFN-β expression; (iii) attenuation of VSV through disruption of normal gene order; (iv) mutating the VSV G protein; (v) introducing targets for microRNA from normal cells into the VSV genome; (vi) pseudotyping VSV; (vii) experimental adaptation of VSV to cancer cells; and (viii) using semi-replicative VSV. Most of the studies in our laboratory focus on VSV-∆M51 recombinants containing a deletion of the methionine residue at position 51 of the M protein, VSV-∆M51. This mutation results in an inability of VSV-M to inhibit nucleus-to-cytoplasm transport of cellular mRNA, including antiviral transcripts, in normal cells with functional antiviral signaling [122][123].
Our laboratory has characterized numerous human PDAC cells lines and discovered a wide range of susceptibility and permissiveness of different PDAC cell lines to VSV and other tested OVs [94][96][98][99][117][124][125]. The range includes “super-permissive” cell lines (such as MIA PaCa-2 and Capan1), “super-resistant” cell lines (such as HPAF-II, Hs766T), and well as many cell lines in between (such as SUIT2 and AsPC-1). Below we describe different mechanisms associated with the resistance of some PDACs to VSV.
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85.
- Aier, I.; Semwal, R.; Sharma, A.; Varadwaj, P.K. A systematic assessment of statistics, risk factors, and underlying features involved in pancreatic cancer. Cancer Epidemiol. 2019, 58, 104–110.
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 1–20.
- Siegel, R.L.; Miller, D.K.; Ahmedi, J. Cancer Facts & Figures 2020. CA Cancer J. Clin. 2020, 70.
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168.
- Salo-Mullen, E.E.; O’Reilly, E.M.; Kelsen, D.P.; Ba, A.M.A.; Lowery, M.A.; Yu, K.H.; Reidy, D.L.; Epstein, A.S.; Lincoln, A.; Bs, A.S.; et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer 2015, 121, 4382–4388.
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients with Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390.
- Yachida, S.; White, C.M.; Naito, Y.; Zhong, Y.; Brosnan, J.A.; Macgregor-Das, A.M.; Morgan, R.A.; Saunders, T.; Laheru, D.A.; Herman, J.M.; et al. Clinical Significance of the Genetic Landscape of Pancreatic Cancer and Implications for Identification of Potential Long-term Survivors. Clin. Cancer Res. 2012, 18, 6339–6347.
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42.
- Wong, W.; Raufi, A.G.; A Safyan, R.; E Bates, S.; A Manji, G. BRCA Mutations in Pancreas Cancer: Spectrum, Current Management, Challenges and Future Prospects. Cancer Manag. Res. 2020, 12, 2731–2742.
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861.
- Springfeld, C.; Jäger, D.; Büchler, M.W.; Strobel, O.; Hackert, T.; Palmer, D.H.; Neoptolemos, J.P. Chemotherapy for pancreatic cancer. Presse Médicale 2019, 48, e159–e174.
- Turpin, A.; El Amrani, M.; Bachet, J.-B.; Pietrasz, D.; Schwarz, L.; Hammel, P. Adjuvant Pancreatic Cancer Management: Towards New Perspectives in 2021. Cancers 2020, 12, 3866.
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.-L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med Oncol. 2019, 11, 1758835919875568.
- Perri, G.; Prakash, L.; Katz, M.H.G. Defining and Treating Borderline Resectable Pancreatic Cancer. Curr. Treat. Options Oncol. 2020, 21, 1–11.
- Parekh, H.D.; Starr, J.; George, T.J. The Multidisciplinary Approach to Localized Pancreatic Adenocarcinoma. Curr. Treat. Options Oncol. 2017, 18, 73.
- Dauer, P.; Nomura, A.; Saluja, A.; Banerjee, S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology 2017, 17, 7–12.
- Kleeff, J.; Beckhove, P.; Esposito, I.; Herzig, S.; Huber, P.E.; Löhr, J.M.; Friess, H. Pancreatic cancer microenvironment. Int. J. Cancer 2007, 121, 699–705.
- Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Razak, N.B.A.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87.
- Akakura, N.; Kobayashi, M.; Horiuchi, I.; Suzuki, A.; Wang, J.; Chen, J.; Niizeki, H.; Ki, K.; Hosokawa, M.; Asaka, M. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001, 61, 6548–6554.
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863.
- Tréhoux, S.; Duchêne, B.; Jonckheere, N.; Van Seuningen, I. The MUC1 oncomucin regulates pancreatic cancer cell biological properties and chemoresistance. Implication of p42–44 MAPK, Akt, Bcl-2 and MMP13 pathways. Biochem. Biophys. Res. Commun. 2015, 456, 757–762.
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495.
- Uchida, D.; Shiraha, H.; Kato, H.; Nagahara, T.; Iwamuro, M.; Kataoka, J.; Horiguchi, S.; Watanabe, M.; Takaki, A.; Nouso, K.; et al. Potential of adenovirus-mediated REIC/Dkk-3 gene therapy for use in the treatment of pancreatic cancer. J. Gastroenterol. Hepatol. 2013, 29, 973–983.
- Horioka, K.; Ohuchida, K.; Sada, M.; Zheng, B.; Moriyama, T.; Fujita, H.; Manabe, T.; Ohtsuka, T.; Shimamoto, M.; Miyazaki, T.; et al. Suppression of CD51 in pancreatic stellate cells inhibits tumor growth by reducing stroma and altering tumor-stromal interaction in pancreatic cancer. Int. J. Oncol. 2016, 48, 1499–1508.
- Barnes, C.A.; Chavez, M.I.; Tsai, S.; Aldakkak, M.; George, B.; Ritch, P.S.; Dua, K.; Clarke, C.N.; Tolat, P.; Hagen, C.; et al. Survival of patients with borderline resectable pancreatic cancer who received neoadjuvant therapy and surgery. Surgery 2019, 166, 277–285.
- Giovannetti, E.; Del Tacca, M.; Mey, V.; Funel, N.; Nannizzi, S.; Ricci, S.; Orlandini, C.; Boggi, U.; Campani, D.; Del Chiaro, M.; et al. Transcription Analysis of Human Equilibrative Nucleoside Transporter-1 Predicts Survival in Pancreas Cancer Patients Treated with Gemcitabine. Cancer Res. 2006, 66, 3928–3935.
- Spratlin, J.; Sangha, R.; Glubrecht, D.; Dabbagh, L.; Young, J.D.; Dumontet, C.; Cass, C.; Lai, R.; Mackey, J.R. The Absence of Human Equilibrative Nucleoside Transporter 1 Is Associated with Reduced Survival in Patients with Gemcitabine-Treated Pancreas Adenocarcinoma. Clin. Cancer Res. 2004, 10, 6956–6961.
- Ohhashi, S.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Egami, T.; Yu, J.; Toma, H.; Sadatomi, S.; Nagai, E.; Tanaka, M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008, 28, 2205–2212.
- Saiki, Y.; Yoshino, Y.; Fujimura, H.; Manabe, T.; Kudo, Y.; Shimada, M.; Mano, N.; Nakano, T.; Lee, Y.; Shimizu, S.; et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 98–104.
- Vernejoul, F.; Ghénassia, L.; Souque, A.; Lulka, H.; Drocourt, D.; Cordelier, P.; Pradayrol, L.; Pyronnet, S.; Buscail, L.; Tiraby, G.; et al. Gene Therapy Based on Gemcitabine Chemosensitization Suppresses Pancreatic Tumor Growth. Mol. Ther. 2006, 14, 758–767.
- Kasuya, K.; Tsuchida, A.; Nagakawa, Y.; Suzuki, M.; Abe, Y.; Itoi, T.; Serizawa, H.; Nagao, T.; Shimazu, M.; Aoki, T. Hypoxia-inducible factor-1α expression and gemcitabine chemotherapy for pancreatic cancer. Oncol. Rep. 2011, 26, 1399–1406.
- Zhang, Z.; Han, H.; Rong, Y.; Zhu, K.; Zhu, Z.; Tang, Z.; Xiong, C.; Tao, J. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J. Exp. Clin. Cancer Res. 2018, 37, 291.
- Wang, S.; You, L.; Dai, M.; Zhao, Y. Mucins in pancreatic cancer: A well-established but promising family for diagnosis, prognosis and therapy. J. Cell. Mol. Med. 2020, 24, 10279–10289.
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e77.
- Mutgan, A.C.; Besikcioglu, H.E.; Wang, S.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol. Cancer 2018, 17, 1–11.
- Friedlander, S.Y.G.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; DePinho, R.A.; Jacks, T. Context-Dependent Transformation of Adult Pancreatic Cells by Oncogenic K-Ras. Cancer Cell 2009, 16, 379–389.
- Kalra, A.V.; Campbell, R.B. Mucin impedes cytotoxic effect of 5-FU against growth of human pancreatic cancer cells: Overcoming cellular barriers for therapeutic gain. Br. J. Cancer 2007, 97, 910–918.
- Nath, S.K.; Daneshvar, K.; Das Roy, L.; Grover, P.L.; Kidiyoor, A.; Mosley, L.J.; Sahraei, M.; Mukherjee, P.K. MUC1 induces drug resistance in pancreatic cancer cells via upregulation of multidrug resistance genes. Oncogenesis 2013, 2, e51.
- Dang, C.V. MUC-king with HIF May Rewire Pyrimidine Biosynthesis and Curb Gemcitabine Resistance in Pancreatic Cancer. Cancer Cell 2017, 32, 3–5.
- Bafna, S.; Kaur, S.; Momi, N.; Batra, S.K. Pancreatic cancer cells resistance to gemcitabine: The role of MUC4 mucin. Br. J. Cancer 2009, 101, 1155–1161.
- Skrypek, N.; Duchêne, B.; Hebbar, M.; Leteurtre, E.; Van Seuningen, I.; Jonckheere, N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene 2012, 32, 1714–1723.
- Mimeault, M.; Johansson, S.L.; Senapati, S.; Momi, N.; Chakraborty, S.; Batra, S.K. MUC4 down-regulation reverses chemoresistance of pancreatic cancer stem/progenitor cells and their progenies. Cancer Lett. 2010, 295, 69–84.
- Amorino, G.P.; Hamilton, V.M.; Valerie, K.; Dent, P.; Lammering, G.; Schmidt-Ullrich, R.K. Epidermal Growth Factor Receptor Dependence of Radiation-induced Transcription Factor Activation in Human Breast Carcinoma Cells. Mol. Biol. Cell 2002, 13, 2233–2244.
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504.
- Khodarev, N.N.; Beckett, M.; Labay, E.; Darga, T.; Roizman, B.; Weichselbaum, R.R. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1714–1719.
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501.
- Eissa, I.R.; Bustos-Villalobos, I.; Ichinose, T.; Matsumura, S.; Naoe, Y.; Miyajima, N.; Morimoto, D.; Mukoyama, N.; Zhiwen, W.; Tanaka, M.; et al. The Current Status and Future Prospects of Oncolytic Viruses in Clinical Trials against Melanoma, Glioma, Pancreatic, and Breast Cancers. Cancers 2018, 10, 356.
- Stevenson, A.J.; Giles, M.S.; Hall, K.T.; Goodwin, D.J.; A Calderwood, M.; Markham, A.F.; Whitehouse, A. Specific oncolytic activity of herpesvirus saimiri in pancreatic cancer cells. Br. J. Cancer 2000, 83, 329–332.
- Kasuya, H.; Nishiyama, Y.; Nomoto, S.; Goshima, F.; Takeda, S.; Watanabe, I.; Nomura, N.; Shikano, T.; Fujii, T.; Kanazumi, N.; et al. Suitability of a US3-inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther. 2007, 14, 533–542.
- Gayral, M.; Lulka, H.; Hanoun, N.; Biollay, C.; Sèlves, J.; Vignolle-Vidoni, A.; Berthommé, H.; Trempat, P.; Epstein, A.L.; Buscail, L.; et al. Targeted Oncolytic Herpes Simplex Virus Type 1 Eradicates Experimental Pancreatic Tumors. Hum. Gene Ther. 2015, 26, 104–113.
- Watanabe, I.; Kasuya, H.; Nomura, N.; Shikano, T.; Shirota, T.; Kanazumi, N.; Takeda, S.; Nomoto, S.; Sugimoto, H.; Nakao, A. Effects of tumor selective replication-competent herpes viruses in combination with gemcitabine on pancreatic cancer. Cancer Chemother. Pharmacol. 2007, 61, 875–882.
- Fu, X.; Tao, L.; Li, M.; Fisher, W.E.; Zhang, X. Effective Treatment of Pancreatic Cancer Xenografts with a Conditionally Replicating Virus Derived from Type 2 Herpes Simplex Virus. Clin. Cancer Res. 2006, 12, 3152–3157.
- Eisenberg, D.P.; Adusumilli, P.S.; Hendershott, K.J.; Yu, Z.; Mullerad, M.; Chan, M.-K.; Chou, T.-C.; Fong, Y. 5-Fluorouracil and Gemcitabine Potentiate the Efficacy of Oncolytic Herpes Viral Gene Therapy in the Treatment of Pancreatic Cancer. J. Gastrointest. Surg. 2005, 9, 1068–1079.
- Liu, H.; Yuan, S.-J.; Chen, Y.-T.; Xie, Y.-B.; Cui, L.; Yang, W.-Z.; Yang, D.-X.; Tian, Y.-T. Preclinical evaluation of herpes simplex virus armed with granulocyte-macrophage colony-stimulating factor in pancreatic carcinoma. World J. Gastroenterol. 2013, 19, 5138–5143.
- Bortolanza, S.; Bunuales, M.; Otano, I.; Gonzalez-Aseguinolaza, G.; Ortiz-De-Solorzano, C.; Perez, D.R.; Prieto, J.; Hernandez-Alcoceba, R. Treatment of Pancreatic Cancer with an Oncolytic Adenovirus Expressing Interleukin-12 in Syrian Hamsters. Mol. Ther. 2009, 17, 614–622.
- McAuliffe, P.F.; Jarnagin, W.R.; Johnson, P.; Delman, K.A.; Federoff, H.; Fong, Y. Effective treatment of pancreatic tumors with two multimutated herpes simplex oncolytic viruses. J. Gastrointest. Surg. 2000, 4, 580–588.
- Nishimoto, T.; Yoshida, K.; Miura, Y.; Kobayashi, A.; Hara, H.; Ohnami, S.; Kurisu, K.; Yoshida, T.; Aoki, K. Oncolytic virus therapy for pancreatic cancer using the adenovirus library displaying random peptides on the fiber knob. Gene Ther. 2009, 16, 669–680.
- Rodríguez-García, A.; Giménez-Alejandre, M.; Rojas, J.J.; Moreno, R.; Bazan-Peregrino, M.; Cascalló, M.; Alemany, R. Safety and Efficacy of VCN-01, an Oncolytic Adenovirus Combining Fiber HSG-Binding Domain Replacement with RGD and Hyaluronidase Expression. Clin. Cancer Res. 2015, 21, 1406–1418.
- Leitner, S.; Sweeney, K.; Öberg, D.; Davies, D.; Miranda, E.; Lemoine, N.R.; Halldén, G. Oncolytic Adenoviral Mutants with E1B19K Gene Deletions Enhance Gemcitabine-induced Apoptosis in Pancreatic Carcinoma Cells and Anti-Tumor Efficacy In Vivo. Clin. Cancer Res. 2009, 15, 1730–1740.
- Armstrong, L.; Arrington, A.; Han, J.; Gavrikova, T.; Brown, E.; Yamamoto, M.; Vickers, S.M.; Davydova, J. Generation of a novel, cyclooxygenase-2–targeted, interferon-expressing, conditionally replicative adenovirus for pancreatic cancer therapy. Am. J. Surg. 2012, 204, 741–750.
- Yu, D.; Jin, C.; Ramachandran, M.; Xu, J.; Nilsson, B.; Korsgren, O.; Le Blanc, K.; Uhrbom, L.; Forsberg-Nilsson, K.; Westermark, B.; et al. Adenovirus Serotype 5 Vectors with Tat-PTD Modified Hexon and Serotype 35 Fiber Show Greatly Enhanced Transduction Capacity of Primary Cell Cultures. PLoS ONE 2013, 8, e54952.
- Zhang, Y.-A.; Nemunaitis, J.; Samuel, S.K.; Chen, P.; Shen, Y.; Tong, A.W. Antitumor Activity of an Oncolytic Adenovirus-Delivered Oncogene Small Interfering RNA. Cancer Res. 2006, 66, 9736–9743.
- Xu, B.; Zheng, W.-Y.; Jin, D.-Y.; Wang, D.-S.; Liu, X.-Y.; Qin, X.-Y. Treatment of pancreatic cancer using an oncolytic virus harboring the lipocalin-2 gene. Cancer 2012, 118, 5217–5226.
- Eriksson, E.; Milenova, I.; Wenthe, J.; Ståhle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin. Cancer Res. 2017, 23, 5846–5857.
- Salzwedel, A.O.; Han, J.; LaRocca, C.J.; Shanley, R.; Yamamoto, M.; Davydova, J. Combination of interferon-expressing oncolytic adenovirus with chemotherapy and radiation is highly synergistic in hamster model of pancreatic cancer. Oncotarget 2018, 9, 18041–18052.
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3.
- Jung, K.H.; Choi, I.-K.; Lee, H.-S.; Yan, H.H.; Son, M.K.; Ahn, H.M.; Hong, J.; Yun, C.-O.; Hong, S.-S. Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett. 2017, 396, 155–166.
- Cherubini, G.; Kallin, C.; Mozetic, A.; Hammaren-Busch, K.; Muller, H.; Lemoine, N.R.; Hallden, G. The oncolytic adenovirus AdDeltaDelta enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011, 18, 1157–1165.
- Bhattacharyya, M.; Francis, J.; Eddouadi, A.; Lemoine, N.R.; Hallden, G. An oncolytic adenovirus defective in pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer cells and tumors in vivo in combination with 5-FU or gemcitabine. Cancer Gene Ther. 2011, 18, 734–743.
- Dai, B.; Roife, D.; Kang, Y.; Gumin, J.; Perez, M.V.R.; Li, X.; Pratt, M.; Brekken, R.A.; Fueyo-Margareto, J.; Lang, F.F.; et al. Preclinical Evaluation of Sequential Combination of Oncolytic Adenovirus Delta-24-RGD and Phosphatidylserine-Targeting Antibody in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 662–670.
- Man, Y.K.S.; Davies, J.A.; Coughlan, L.; Pantelidou, C.; Blazquez-Moreno, A.; Marshall, J.F.; Parker, A.L.; Hallden, G. The Novel Oncolytic Adenoviral Mutant Ad5-3Delta-A20T Retargeted to alphavbeta6 Integrins Efficiently Eliminates Pancreatic Cancer Cells. Mol. Cancer Ther. 2018, 17, 575–587.
- Rovira-Rigau, M.; Raimondi, G.; Marín, M.Á.; Gironella, M.; Alemany, R.; Fillat, C. Bioselection Reveals miR-99b and miR-485 as Enhancers of Adenoviral Oncolysis in Pancreatic Cancer. Mol. Ther. 2019, 27, 230–243.
- Freytag, S.O.; Barton, K.N.; Brown, S.L.; Narra, V.; Zhang, Y.; Tyson, D.; Nall, C.; Lü, M.; Ajlouni, M.; Movsas, B.; et al. Replication-competent Adenovirus-mediated Suicide Gene Therapy with Radiation in a Preclinical Model of Pancreatic Cancer. Mol. Ther. 2007, 15, 1600–1606.
- Woo, Y.; Kelly, K.J.; Stanford, M.M.; Galanis, C.; Chun, Y.S.; Fong, Y.; McFadden, G. Myxoma Virus Is Oncolytic for Human Pancreatic Adenocarcinoma Cells. Ann. Surg. Oncol. 2008, 15, 2329–2335.
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Kelly, K.R.; Coffey, M.; Freeman, J.W.; Nawrocki, S.T. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013, 4, e728.
- Chen, W.; Fan, W.; Ru, G.; Huang, F.; Lu, X.; Zhang, X.; Mou, X.; Wang, S. Gemcitabine combined with an engineered oncolytic vaccinia virus exhibits a synergistic suppressive effect on the tumor growth of pancreatic cancer. Oncol. Rep. 2018, 41, 67–76.
- Sun, T.; Luo, Y.; Wang, M.; Xie, T.; Yan, H. Recombinant Oncolytic Vaccinia Viruses Expressing Human beta-Defensin 2 Enhance Anti-tumor Immunity. Mol. Ther. Oncolytics 2019, 13, 49–57.
- Wu, Y.; Mou, X.; Wang, S.; Liu, X.-E.; Sun, X. ING4 expressing oncolytic vaccinia virus promotes anti-tumor efficiency and synergizes with gemcitabine in pancreatic cancer. Oncotarget 2017, 8, 82728–82739.
- Chard, L.S.; Lemoine, N.R.; Wang, Y. New role of Interleukin-10 in enhancing the antitumor efficacy of oncolytic vaccinia virus for treatment of pancreatic cancer. OncoImmunology 2015, 4, e1038689.
- Tysome, J.R.; Briat, A.; Alusi, G.; Cao, F.; Gao, D.; Yu, J.; Wang, P.; Yang, S.; Dong, Z.; Wang, S.; et al. Lister strain of vaccinia virus armed with endostatin–angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 2009, 16, 1223–1233.
- Bhat, R.; Dempe, S.; Dinsart, C.; Rommelaere, J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int. J. Cancer 2010, 128, 908–919.
- Moehler, M.; Sieben, M.; Roth, S.; Springsguth, F.; Leuchs, B.; Zeidler, M.; Dinsart, C.; Rommelaere, J.; Galle, P.R. Activation of the human immune system by chemotherapeutic or targeted agents combined with the oncolytic parvovirus H-1. BMC Cancer 2011, 11, 464.
- Li, J.; Bonifati, S.; Hristov, G.; Marttila, T.; Valmary-Degano, S.; Stanzel, S.; Schnölzer, M.; Mougin, C.; Aprahamian, M.; Grekova, S.P.; et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1 PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol. Med. 2013, 5, 1537–1555.
- Bossow, S.; Grossardt, C.; Temme, A.; Leber, M.F.; Sawall, S.; Rieber, E.P.; Cattaneo, R.; Von Kalle, C.; Ungerechts, G. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011, 18, 598–608.
- May, V.; Berchtold, S.; Berger, A.; Venturelli, S.; Burkard, M.; Leischner, C.; Malek, N.P.; Lauer, U.M. Chemovirotherapy for pancreatic cancer: Gemcitabine plus oncolytic measles vaccine virus. Oncol. Lett. 2019, 18, 5534–5542.
- Walter, R.J.; Attar, B.M.; Rafiq, A.; Tejaswi, S.; Delimata, M. Newcastle disease virus LaSota strain kills human pancreatic cancer cells in vitro with high selectivity. JOP 2012, 13, 45–53.
- Schwaiger, T.; Knittler, M.R.; Grund, C.; Roemer-Oberdoerfer, A.; Kapp, J.-F.; Lerch, M.M.; Mettenleiter, T.C.; Mayerle, J.; Blohm, U. Newcastle disease virus mediates pancreatic tumor rejection via NK cell activation and prevents cancer relapse by prompting adaptive immunity. Int. J. Cancer 2017, 141, 2505–2516.
- Fábián, Z.; Csatary, C.M.; Szeberényi, J.; Csatary, L.K. p53-Independent Endoplasmic Reticulum Stress-Mediated Cytotoxicity of a Newcastle Disease Virus Strain in Tumor Cell Lines. J. Virol. 2007, 81, 2817–2830.
- O’Leary, M.P.; Choi, A.H.; Kim, S.-I.; Chaurasiya, S.; Lu, J.; Park, A.K.; Woo, Y.; Warner, S.G.; Fong, Y.; Chen, N.G. Novel oncolytic chimeric orthopoxvirus causes regression of pancreatic cancer xenografts and exhibits abscopal effect at a single low dose. J. Transl. Med. 2018, 16, 1–11.
- Kasloff, S.B.; Pizzuto, M.S.; Silic-Benussi, M.; Pavone, S.; Ciminale, V.; Capua, I. Oncolytic Activity of Avian Influenza Virus in Human Pancreatic Ductal Adenocarcinoma Cell Lines. J. Virol. 2014, 88, 9321–9334.
- Blackham, A.U.; Northrup, S.A.; Willingham, M.; Sirintrapun, J.; Russell, G.B.; Lyles, D.S.; Stewart, J.H. Molecular determinants of susceptibility to oncolytic vesicular stomatitis virus in pancreatic adenocarcinoma. J. Surg. Res. 2014, 187, 412–426.
- Nagalo, B.M.; Breton, C.A.; Zhou, Y.; Arora, M.; Bogenberger, J.M.; Barro, O.; Steele, M.B.; Jenks, N.J.; Baker, A.T.; Duda, D.G.; et al. Oncolytic Virus with Attributes of Vesicular Stomatitis Virus and Measles Virus in Hepatobiliary and Pancreatic Cancers. Mol. Ther. Oncolytics 2020, 18, 546–555.
- Murphy, A.M.; Besmer, D.M.; Moerdyk-Schauwecker, M.; Moestl, N.; Ornelles, D.A.; Mukherjee, P.; Grdzelishvili, V.Z. Vesicular Stomatitis Virus as an Oncolytic Agent against Pancreatic Ductal Adenocarcinoma. J. Virol. 2012, 86, 3073–3087.
- Heiber, J.F.; Barber, G.N. Vesicular Stomatitis Virus Expressing Tumor Suppressor p53 Is a Highly Attenuated, Potent Oncolytic Agent. J. Virol. 2011, 85, 10440–10450.
- Bressy, C.; Droby, G.N.; Maldonado, B.D.; Steuerwald, N.; Grdzelishvili, V.Z. Cell Cycle Arrest in G2/M Phase Enhances Replication of Interferon-Sensitive Cytoplasmic RNA Viruses via Inhibition of Antiviral Gene Expression. J. Virol. 2018, 93, e01885-18.
- Gaddy, D.F.; Lyles, D.S. Vesicular Stomatitis Viruses Expressing Wild-Type or Mutant M Proteins Activate Apoptosis through Distinct Pathways. J. Virol. 2005, 79, 4170–4179.
- Cataldi, M.; Shah, N.R.; Felt, S.A.; Grdzelishvili, V.Z. Breaking resistance of pancreatic cancer cells to an attenuated vesicular stomatitis virus through a novel activity of IKK inhibitor TPCA-1. Virology 2015, 485, 340–354.
- Felt, S.A.; Droby, G.N.; Grdzelishvili, V.Z. Ruxolitinib and Polycation Combination Treatment Overcomes Multiple Mechanisms of Resistance of Pancreatic Cancer Cells to Oncolytic Vesicular Stomatitis Virus. J. Virol. 2017, 91, 91.
- Hastie, E.; Besmer, D.M.; Shah, N.R.; Murphy, A.M.; Moerdyk-Schauwecker, M.; Molestina, C.; Das Roy, L.; Curry, J.M.; Mukherjee, P.; Grdzelishvili, V.Z. Oncolytic Vesicular Stomatitis Virus in an Immunocompetent Model of MUC1-Positive or MUC1-Null Pancreatic Ductal Adenocarcinoma. J. Virol. 2013, 87, 10283–10294.
- Seegers, S.L.; Frasier, C.; Greene, S.; Nesmelova, I.V.; Grdzelishvili, V.Z. Experimental Evolution Generates Novel Oncolytic Vesicular Stomatitis Viruses with Improved Replication in Virus-Resistant Pancreatic Cancer Cells. J. Virol. 2019, 94, 94.
- Hecht, J.R.; Bedford, R.; Abbruzzese, J.L.; Lahoti, S.; Reid, T.R.; Soetikno, R.M.; Kirn, D.H.; Freeman, S.M. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin. Cancer Res. 2003, 9, 555–561.
- Mulvihill, S.; Warren, R.; Venook, A.; Adler, A.; Randlev, B.; Heise, C.; Kirn, D. Safety and feasibility of injection with an E1B-55 kDa gene-deleted, replication-selective adenovirus (ONYX-015) into primary carcinomas of the pancreas: A phase I trial. Gene Ther. 2001, 8, 308–315.
- Lee, J.-C.; Shin, D.W.; Park, H.; Kim, J.; Youn, Y.; Kim, J.H.; Kim, J.; Hwang, J.-H. Tolerability and safety of EUS-injected adenovirus-mediated double-suicide gene therapy with chemotherapy in locally advanced pancreatic cancer: A phase 1 trial. Gastrointest. Endosc. 2020, 92, 1044–1052.e1.
- Chang, K.J.; Senzer, N.N.; Binmoeller, K.; Goldsweig, H.; Coffin, R. Phase I dose-escalation study of talimogene laherparepvec (T-VEC) for advanced pancreatic cancer (ca). J. Clin. Oncol. 2012, 30, e14546.
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; Naoe, Y.; Ichinose, T.; Koyama, N.; Tanaka, M.; et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer 2018, 18, 1–9.
- Noonan, A.M.; Farren, M.R.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized Phase 2 Trial of the Oncolytic Virus Pelareorep (Reolysin) in Upfront Treatment of Metastatic Pancreatic Adenocarcinoma. Mol. Ther. 2016, 24, 1150–1158.
- Mahalingam, D.; Goel, S.; Aparo, S.; Patel Arora, S.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Gutierrez, A.; Coffey, M.; et al. A Phase II Study of Pelareorep (REOLYSIN((R))) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers 2018, 10, 160.
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.H.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in Combination with the Oncolytic Virus Pelareorep and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma: A Phase Ib Study. Clin. Cancer Res. 2019, 26, 71–81.
- Hajda, J.; Lehmann, M.; Krebs, O.; Kieser, M.; Geletneky, K.; Jäger, D.; Dahm, M.; Huber, B.; Schöning, T.; Sedlaczek, O.; et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer 2017, 17, 1–11.
- Monsurrò, V.; Beghelli, S.; Wang, R.; Barbi, S.; Coin, S.; Di Pasquale, G.; Bersani, S.; Castellucci, M.; Sorio, C.; Eleuteri, S.; et al. Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: Potential relevance for adenoviral gene therapy. J. Transl. Med. 2010, 8, 10.
- Bressy, C.; Hastie, E.; Grdzelishvili, V.Z. Combining Oncolytic Virotherapy with p53 Tumor Suppressor Gene Therapy. Mol. Ther. Oncolytics 2017, 5, 20–40.
- Wennier, S.T.; Liu, J.; Li, S.; Rahman, M.M.; Mona, M.; McFadden, G. Myxoma Virus Sensitizes Cancer Cells to Gemcitabine and Is an Effective Oncolytic Virotherapeutic in Models of Disseminated Pancreatic Cancer. Mol. Ther. 2012, 20, 759–768.
- Hastie, E.; Grdzelishvili, V.Z. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 2012, 93, 2529–2545.
- Hastie, E.; Cataldi, M.; Marriott, I.; Grdzelishvili, V.Z. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res. 2013, 176, 16–32.
- Felt, S.A.; Grdzelishvili, V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: A 5-year update. J. Gen. Virol. 2017, 98, 2895–2911.
- Moerdyk-Schauwecker, M.; Shah, N.R.; Murphy, A.M.; Hastie, E.; Mukherjee, P.; Grdzelishvili, V.Z. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: Role of type I interferon signaling. Virology 2013, 436, 221–234.
- Moussavi, M.; Fazli, L.; Tearle, H.; Guo, Y.; Cox, M.; Bell, J.; Ong, C.; Jia, W.; Rennie, P.S. Oncolysis of Prostate Cancers Induced by Vesicular Stomatitis Virus in PTEN Knockout Mice. Cancer Res. 2010, 70, 1367–1376.
- Zhang, K.-X.; Matsui, Y.; Hadaschik, B.A.; Lee, C.; Jia, W.; Bell, J.C.; Fazli, L.; So, A.I.; Rennie, P.S. Down-regulation of type I interferon receptor sensitizes bladder cancer cells to vesicular stomatitis virus-induced cell death. Int. J. Cancer 2010, 127, 830–838.
- Noser, J.A.; Mael, A.A.; Sakuma, R.; Ohmine, S.; Marcato, P.; Lee, P.W.; Ikeda, Y. The RAS/Raf1/MEK/ERK Signaling Pathway Facilitates VSV-mediated Oncolysis: Implication for the Defective Interferon Response in Cancer Cells. Mol. Ther. 2007, 15, 1531–1536.
- Li, Q.; Tainsky, M.A. Epigenetic Silencing of IRF7 and/or IRF5 in Lung Cancer Cells Leads to Increased Sensitivity to Oncolytic Viruses. PLoS ONE 2011, 6, e28683.
- Stojdl, D.F.; Lichty, B.D.; Tenoever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275.
- Petersen, J.M.; Her, L.-S.; Varvel, V.; Lund, E.; Dahlberg, J.E. The Matrix Protein of Vesicular Stomatitis Virus Inhibits Nucleocytoplasmic Transport When It Is in the Nucleus and Associated with Nuclear Pore Complexes. Mol. Cell. Biol. 2000, 20, 8590–8601.
- Hastie, E.; Cataldi, M.; Moerdyk-Schauwecker, M.J.; Felt, S.A.; Steuerwald, N.; Grdzelishvili, V.Z. Novel biomarkers of resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus. Oncotarget 2016, 7, 61601–61618.
- Felt, S.A.; Moerdyk-Schauwecker, M.J.; Grdzelishvili, V.Z. Induction of apoptosis in pancreatic cancer cells by vesicular stomatitis virus. Virology 2015, 474, 163–173.


