 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sara Lemoinne | + 1619 word(s) | 1619 | 2021-04-08 07:58:20 | | | |
| 2 | Dean Liu | -10 word(s) | 1609 | 2021-04-19 05:17:32 | | |
Video Upload Options
Non-alcoholic fatty liver disease (NAFLD) has become the leading cause of chronic liver disease, exposing to the risk of liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC).
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) has become the most common cause of chronic liver disease worldwide [1]. NAFLD includes non-alcoholic fatty liver (NAFL) defined by steatosis and non-alcoholic steatohepatitis (NASH) defined by steatosis associated with inflammation and hepatocyte ballooning. NASH can progress to fibrosis, cirrhosis and hepatocellular carcinoma (HCC), accounting for a significant health burden. However, in spite of its high prevalence and significant health burden, there is currently no approved pharmacological therapy to treat patients with NAFLD.
Angiogenesis is a complex process leading to the development of new vessels from pre-existing vessels. Angiogenesis occurs under physiological conditions during normal wound healing and also in pathological contexts, such as tumorigenesis, so that antiangiogenic molecules (e.g., of Bevacizumab, an anti-vascular endothelial growth factor (VEGF) monoclonal antibody) are used in the treatment of different cancers, including HCC, according to recent guidelines [2]. Molecular and cellular mechanisms of angiogenesis have been extensively studied and are explained in detail elsewhere [3]. In short, in the quiescent state, endothelial cells are organized in a monolayer of cells, connected by intercellular junctions and surrounded by pericytes, which control their proliferation via the secretion of survival signals, such as VEGF and angiopoietin-1. When a vessel receives a proangiogenic signal such as VEGF, angiopoietin-2 (Ang-2), fibroblast growth factor (FGF) or another chemokine secreted by a hypoxic cell or an inflammatory cell, first, peri-cytes detach from the vessel in response to Ang-2. Then, endothelial cells become activated, lose their intercellular junctions and proliferate. VEGF increases the endothelial permeability, leading to the extravasation of plasma proteins which will make the scaffold of a temporary extracellular matrix. Endothelial cells migrate on the surface of this new extracellular matrix, forming a stalk that progressively elongates to build a new vessel. Once the neovessel is built, a maturation step is required to make the vessel functional. During this maturation step, endothelial cells go back to a quiescent state and pericytes are recruited to surround the endothelial cells, under the action of platelet-derived growth factor (PDGF), angiopoietin-1, transforming growth factor-ß (TGF-ß) and Notch. VEGF, the main proangiogenic cytokine, plays a central role in angiogenesis since it is implicated in all steps of angiogenesis: VEGF increases vascular permeability, induces endothelial cell proliferation and regulates neovessel lumen diameter [3]. Angiogenic effects of VEGF are mediated via its receptor VEGF receptor-2 (VEGFR-2), which is a tyrosine kinase receptor, expressed at the surface of endothelial cells [3].
Angiogenesis also takes place during chronic liver diseases. Indeed, liver fibrosis progression is accompanied by angiogenesis, regardless of the etiology of the liver disease [4][5]. In this setting, angiogenesis is triggered by hypoxia and inflammation, and its main effect is the aggravation of liver fibrosis, leading to cirrhosis [6].
In the context of chronic liver disease, angiogenesis leads to quantitative changes of liver vessels with the emergence of new vessels but also consists in qualitative changes of vessels (both pre-existing and new vessels), resulting in a process known as vascular remodeling. Such qualitative vascular changes include dedifferentiation of liver sinusoidal endothelial cells (LSECs), also called capillarization, defined by the loss of their fenestrae and the acquisition of a basement membrane [7]. In the studies of angiogenesis during chronic liver disease, it is particularly difficult to discriminate LSECs from vascular endothelial cells in the liver, first because not a single marker is fully specific of LSECs, and also because LSECs lose the expression of their canonical markers when they undergo capillarization [7].
2. Role of Angiogenesis in the Progression of NAFLD
Evidence of liver angiogenesis in animal models of NAFLD and in NAFLD patients suggests a role of liver angiogenesis in NAFLD pathogenesis. During NAFLD, angiogenesis drives inflammation and fibrosis, as reviewed elsewhere [8]. Liver angiogenesis is not a specific event in NAFLD as it occurs in all chronic liver diseases with the progression of liver fibrosis [4][5][9]. However, specific features of angiogenesis have been reported in NAFLD. First, the upregulation of proangiogenic genes occurs very early during NAFLD progression, at the stage of pure steatosis, before NASH development, suggesting that angiogenesis is an early event during the physiopathology of NAFLD [10]. Steatosis by itself is able to induce hypoxia, which is the primary inducer of VEGF and trigger of angiogenesis. Indeed, steatosis by boosting the metabolism of fatty acids increases oxygen consumption, generating a hypoxic proangiogenic micro-environment. Steatosis also causes mechanical pressure on sinusoids [11], which further aggravates the shortage of oxygen supply, explaining why patients with pure steatosis may develop portal hypertension [12]. These two mechanisms probably lead to hypoxia and explain why steatosis by itself induces the expression of VEGF [13]. Moreover, hypoxia could be more critical in NAFLD compared to other chronic liver diseases since injury in NAFLD primarily occurs in the perivenular zone, which is more susceptible to hypoxia than the periportal zone, that is primarily injured in other types of liver diseases such as viral hepatitis or biliary diseases. Finally, liver angiogenesis mainly involves LSECs [10], which play a key role in NAFLD pathogenesis, as reviewed elsewhere [14]. For all these reasons, one may infer that angiogenesis is particularly intense in NAFLD, although a comparison of angiogenesis in NAFLD versus other chronic liver diseases, either in animal models or in patients, is lacking.
The molecular pathways of angiogenesis are intermingled with those of NAFLD. Proangiogenic cytokines have an impact on NAFLD. Indeed, VEGF is also involved in lipogenesis so that anti-VEGFR2 treatment can induce changes in the expression of lipogenesis genes, as shown in MCD mice [10]. Hypoxia-inducible factors (HIFs), master effectors of hypoxia, also regulate the expression of genes involved in glucose metabolism [15]. Conversely, cytokines involved in NAFLD development have an impact on angiogenesis. For instance, leptin, an adipokine, which regulates satiety with a key role in obesity, has been shown to stimulate angiogenesis [16]. Kitade et al. showed that leptin-deficient rats exposed to a CDAA diet developed NASH but without neovascularization, as opposed to wild-type rats, clearly indicating that leptin is necessary for angiogenesis in NAFLD [17]. Another example is TGF-β, a master profibrogenic cytokine, which promotes the activation of liver myofibroblasts [18]. TGF-β has also proangiogenic capacities, notably by positively regulating the pericyte differentiation, proliferation and migration [19]. TGF-β is upregulated in NAFLD patients [20] and is increased in patients with NASH compared to patients with pure steatosis [21]. Angiotensin II is known to have profibrogenic properties [22] and angiotensin II-mediated signaling is involved at multiple levels in the development and progression of NAFLD [23]. Angiotensin II has also proangiogenic capacities. Particularly, angiotensin II induced the expression of VEGF in hepatic stellate cells [24].
Increasing evidence indicates that macrophages play a critical role in NAFLD development and progression [25][26]. Yet, macrophages also exert proangiogenic properties in chronic liver diseases. RNA sequencing analysis showed that macrophages isolated from the liver of MCD mice (especially monocyte-derived macrophages) expressed not only inflammatory cytokines but also growth factors involved in angiogenesis [27]. Furthermore, Miura et al. demonstrated that the macrophages were the source of VEGF production in steatotic livers from MCD mice [13]. Yet, in non-NAFLD animal models of chronic liver disease, monocytes-derived macrophages also accumulate in injured livers and exhibit proangiogenic gene profiles, including upregulated VEGF expression [28]. In this latter study, the inhibition of monocyte infiltration prevented angiogenesis but not fibrosis progression, demonstrating the direct role of macrophages in angiogenesis and no strict dependence between angiogenesis on one hand and liver fibrosis on the other [28].
One of the mechanisms that promote angiogenesis is the secretion of extracellular vesicles. Extracellular vesicles are submicron membrane-bound structures secreted from different cell types. They contain a wide variety of molecules and exert important functions in cell-to-cell communication [29]. Many studies have reported the angiogenic pro-perties of extracellular vesicles in different settings [9][30]. Extracellular vesicles are also implicated in the pathophysiology of liver diseases [31][32]. Povero et al. showed that hepatocytes exposed to free fatty acids in vitro released extracellular vesicles able to induce angiogenesis, both in vitro and in vivo, via vanin-1-dependent mechanisms [33]. In this study, the authors observed high levels of circulating hepatocyte-derived extracellular vesicles that were associated with marked liver angiogenesis in mice fed with an MCD diet [33]. Mechanisms promoting angiogenesis in NAFLD are illustrated in Figure 1.
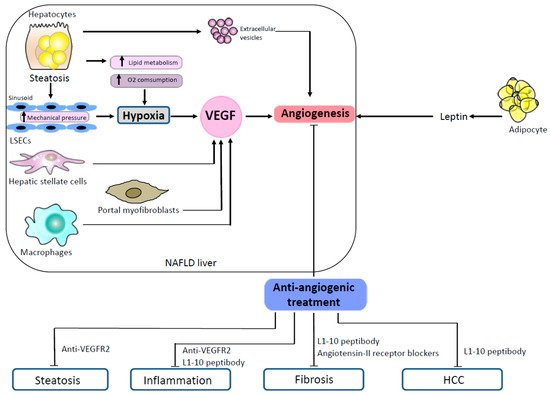
Figure 1. Mechanisms promoting angiogenesis in NAFLD and effects of antiangiogenic treatments in animal models. In NAFLD, steatotic hepatocytes produce proangiogenic extracellular vesicles. Steatosis induces hypoxia both by an increased lipid metabolism which enhances oxygen consumption and by a mechanical pressure on sinusoids. Hepatic stellate cells, portal myofibroblasts and macrophages stimulate angiogenesis by secreting VEGF. Proangiogenic signals also come from the adipose tissue which secretes leptin. In animal models of NAFLD, several antiangiogenic treatments (anti-VEGR2, L1-10 peptibody, angiotensin II receptor blockers) have shown efficacy to reduce steatosis, inflammation, fibrosis and HCC. Abbreviations: HCC, hepatocellular carcinoma; LSECs, liver sinusoidal endothelial cells; NAFLD, non-alcoholic fatty liver disease; VEGF, vascular endothelial growth factor; VEGFR-2, vascular endothelial growth factor receptor-2.
Cirrhosis is associated with the risk of developing HCC in all chronic liver diseases including NAFLD. However, non-cirrhotic patients with NASH have a higher risk of HCC compared to patients with other types of liver disease [34]. HCC is a highly vascularized tumor, a feature that is exploited in the imaging-based diagnosis of HCC. Angiogenesis is a key driver of HCC, and the tumoral expression of angiogenic factors, notably Ang-2, is associated with a pejorative prognosis in HCC [35]. As mentioned earlier, Ang-2 is overexpressed in the liver of patients with NAFLD [36]. Therefore, more sustained angiogenesis could contribute to a higher risk of developing HCC in NAFLD.
Ultimately, the best way to demonstrate the role of angiogenesis in the pathogenesis of NAFLD is to assess the effect of antiangiogenic treatments in NAFLD.
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Gordan, J.D.; Kennedy, E.B.; Abou-Alfa, G.K.; Beg, M.S.; Brower, S.T.; Gade, T.P.; Goff, L.; Gupta, S.; Guy, J.; Harris, W.P.; et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 4317–4345.
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nat. Cell Biol. 2011, 473, 298–307.
- Coulon, S.; Heindryckx, F.; Geerts, A.; Van Steenkiste, C.; Colle, I.; Van Vlierberghe, H. Angiogenesis in chronic liver disease and its complications. Liver Int. 2010, 31, 146–162.
- Zadorozhna, M.; Di Gioia, S.; Conese, M.; Mangieri, D. Neovascularization is a key feature of liver fibrosis progression: Anti-angiogenesis as an innovative way of liver fibrosis treatment. Mol. Biol. Rep. 2020, 47, 2279–2288.
- Lemoinne, S.; Thabut, M.; Housset, C. Portal myofibroblasts connect angiogenesis and fibrosis in liver. Cell Tissue Res. 2016, 365, 583–589.
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227.
- Lefere, S.; Devisscher, L.; Geerts, A. Angiogenesis in the progression of non-alcoholic fatty liver disease. Acta Gastroenterol. Belg. 2020, 83, 301–307.
- Lemoinne, S.; Cadoret, A.; Rautou, P.; El Mourabit, H.; Ratziu, V.; Corpechot, C.; Rey, C.; Bosselut, N.; Barbu, V.; Wendum, D.; et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology 2015, 61, 1041–1055.
- Coulon, S.; Legry, V.; Heindryckx, F.; Van Steenkiste, C.; Casteleyn, C.; Olievier, K.; Libbrecht, L.; Carmeliet, P.; Jonckx, B.; Stassen, J.-M.; et al. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology 2013, 57, 1793–1805.
- Farrell, G.C.; Teoh, N.; McCuskey, R. Hepatic Microcirculation in Fatty Liver Disease. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2008, 291, 684–692.
- Baffy, G. Origins of Portal Hypertension in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2018, 63, 563–576.
- Miura, K.; Ohnishi, H.; Morimoto, N.; Minami, S.; Ishioka, M.; Watanabe, S.; Tsukui, M.; Takaoka, Y.; Nomoto, H.; Isoda, N.; et al. Ezetimibe suppresses development of liver tumors by inhibiting angiogenesis in mice fed a high-fat diet. Cancer Sci. 2018, 110, 771–783.
- Hammoutene, A.; Rautou, P.-E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291.
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2019, 15, 21–32.
- Khazaei, M.; Tahergorabi, Z. Leptin and its cardiovascular effects: Focus on angiogenesis. Adv. Biomed. Res. 2015, 4, 79.
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology 2006, 44, 983–991.
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419.
- Van Meeteren, L.A.; Goumans, M.J.; ten Dijke, P. TGF-beta receptor signaling pathways in angiogenesis; emerging targets for anti-angiogenesis therapy. Curr. Pharm. Biotechnol. 2011, 12, 2108–2120.
- Das, S.K.; Balakrishnan, V. Role of Cytokines in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Indian J. Clin. Biochem. 2011, 26, 202–209.
- Hasegawa, T.; Yoneda, M.; Nakamura, K.; Makino, I.; Terano, A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: A pilot study. Aliment Pharmacol. Ther. 2001, 15, 1667–1672.
- Granzow, M.; Schierwagen, R.; Klein, S.; Kowallick, B.; Huss, S.; Linhart, M.; Mazar, I.G.R.; Görtzen, J.; Vogt, A.; Schildberg, F.A.; et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology 2014, 60, 334–348.
- Morris, E.M.; Fletcher, J.A.; Thyfault, J.P.; Rector, R.S. The role of angiotensin II in nonalcoholic steatohepatitis. Mol. Cell Endocrinol. 2013, 378, 29–40.
- Yoshiji, H.; Kuriyama, S.; Noguchi, R.; Ikenaka, Y.; Kitade, M.; Kaji, K.; Yoshii, J.; Yanase, K.; Yamazaki, M.; Asada, K. Angiotensin-II and vascular endothelial growth factor interaction plays an important role in rat liver fibrosis development. Hepatol. Res. 2006, 36, 124–129.
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43.
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nat. Cell Biol. 2019, 575, 512–518.
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283.
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971.
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705.
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular Vesicles in Angiogenesis. Circ. Res. 2017, 120, 1658–1673.
- Lemoinne, S.; Thabut, D.; Housset, C.; Moreau, R.; Valla, D.; Boulanger, C.M.; Rautou, P.-E. The emerging roles of microvesicles in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 350–361.
- Szabo, G.; Momen-Heravi, F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 455–466.
- Povero, D.; Eguchi, A.; Niesman, I.R.; Andronikou, N.; Jeu, X.D.M.D.; Mulya, A.; Berk, M.; Lazic, M.; Thapaliya, S.; Parola, M.; et al. Lipid-Induced Toxicity Stimulates Hepatocytes to Release Angiogenic Microparticles That Require Vanin-1 for Uptake by Endothelial Cells. Sci. Signal. 2013, 6, ra88.
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703.
- Villa, E.; Critelli, R.; Lei, B.; Marzocchi, G.; Cammà, C.; Giannelli, G.; Pontisso, P.; Cabibbo, G.; Enea, M.; Colopi, S.; et al. Neoangiogenesis-related genes are hallmarks of fast-growing hepatocellular carcinomas and worst survival. Results from a prospective study. Gut 2016, 65, 861–869.
- Lefere, S.; Van De Velde, F.; Hoorens, A.; Raevens, S.; Van Campenhout, S.; Vandierendonck, A.; Neyt, S.; Vandeghinste, B.; Vanhove, C.; Debbaut, C.; et al. Angiopoietin-2 Promotes Pathological Angiogenesis and Is a Therapeutic Target in Murine Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1087–1104.


