 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natalia A. Shnayder | + 2691 word(s) | 2691 | 2021-04-14 10:32:01 | | | |
| 2 | Camila Xu | Meta information modification | 2691 | 2021-04-15 08:13:50 | | | | |
| 3 | Camila Xu | Meta information modification | 2691 | 2021-10-11 03:38:58 | | |
Video Upload Options
Arterial hypertension (AH) is a prevalent condition worldwide and is the key risk factor for non-fatal and fatal cardiovascular complications. Tension-type headache (TTH) is the most common type of primary headache and is considered a common everyday headache.
1. Introduction
Arterial hypertension (AH) is a prevalent condition worldwide and is the key risk factor for non-fatal and fatal cardiovascular complications [1]. Tension-type headache (TTH) is the most common type of primary headache and is considered a common everyday headache [2]. Many studies support the hypothesis that TTH patients have an increased risk of developing AH, while hypertensive subjects do seem to have an increased risk of TTH. The relationship between AH and TTH is potentially of great pathophysiological and clinical interest, but it is poorly understood [3]. This allows us to hypothesize the presence of the AH and TTH phenotype.
The pathophysiological patterns are significantly different in the setting of chronic pain, in which the adaptive relationship between blood pressure and pain sensitivity is reversed. The connection between acute or chronic pain and cardiovascular changes is supported observationally [4], but some of this indirect evidence is confirmed by experimental animal models and human studies. AH and TTH may share common mechanisms like endothelial dysfunction, deficiency of autonomic cardiovascular regulation, and renin angiotensin system involvement.
Nitric oxide (NO) is an important autocrine and paracrine signaling molecule that plays a crucial role in cardiovascular physiology and pathology regulation. NO is an important molecule involved in regulation of cerebral and extra cerebral cranial blood flow and arterial diameters [5]. Reduced bioavailability of NO in the endothelium is an important precursor for impaired vasodilation and AH. Furthermore, NO is involved in nociceptive processing. A NO-induced biphasic response with an immediate and a delayed headache is common to chronic TTH in humans [6].
NO-synthases (NOSs) are expressed in three isoforms, including neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3) [7]. All NOS isoforms can catalyze the conversion of l-arginine to L-citrulline and NO (Figure 1). The active NOSs form a homodimer and convert the amino acid L-arginine to L-citrulline and NO. The NOS monomer contains a C-terminal reductase domain and a N-terminal oxygenase domain, which are linked by the calmodulin (CaM) binding region. The N-terminal oxidase domain contains the heme, tetrahydrobiopterin cofactors (BH4), and the binding site for the substrate arginine. The oxidase domain is the active site of NO synthesis. Production of NO requires O2 as an electron acceptor. NO diffuses freely across the plasma membrane. Therefore, NO can be transported to effector proteins in the adjacent cells and exert its effects (e.g., endothelial NO targets soluble guanylate cyclase (sGC) in smooth muscle to accomplish vasodilation) [8].
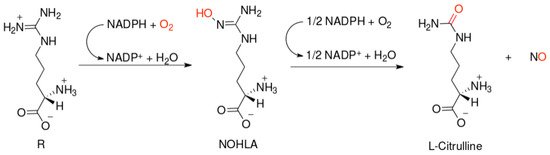
Figure 1. Scheme of nitric oxide (NO) formation from L-arginine in humans.
NOS1 and NOS3 are commonly associated with low levels of NO production, which mediates intracellular signaling processes (NOS1) and vascular homeostasis (NOS3). In addition to NO production, eNOS can function in an uncoupled manner and produce ROS when the available stores of BH4 are removed or oxidized, l-arginine is depleted, or the eNOS inhibitor asymmetric dimethyl-l-arginine is overexpressed [9][10]. nNOS and eNOS are most commonly found in non-immunological cells (e.g., neurons, muscle, endothelium). As their NO output is relatively low, these isoforms are considered less immunologically important than their inducible counterpart, iNOS [7].
NO and NOSs play an important role in the pathogenesis and treatment of both AH and TTH. In particular, NOSs affect the sensitivity of the brain to stress and changes in systemic and local hemodynamics [11]. In addition, a NO-synthesis change in endothelial cells of peripheral and cerebral vessels is one of the identified causes of NO-dependent vasospasm (impaired NO-dependent vasodilation) in patients with AH [12] and TTH [13], as well as chronic pain syndrome [13][14] and changes in the response to antihypertensive drugs [15] and analgesics [16][17]. However, the functional activity of enzymes of the NOS family depends on the carriage of wild, highly functional, low functional, and non-functional single nucleotide variants (SNVs) of NOS1, NOS2, and NOS3 genes encoding the enzyme isoforms.
2. Role of NO and NOSs in Pathogenesis and Treatment of AH
AH is characterized by endothelial dysfunction, vasoconstriction and microvascular rarefaction. All of these mechanisms are interconnected by cause and effect relationships. Moreover, NO is involved in each of these pathogenetic mechanisms.
Thus, NO is the main mediator of endothelial dysfunction underlying the development of AH. Endothelial dysfunction is the first step in the development of atherosclerosis; it is characterized by reduced biosynthesis and decreased bioavailability of NO [11]. NO is the active radical form of both oxygen (ROS) and nitrogen (RNS). These compounds can participate in free radical chain reactions or damage organic substrates. Chain reactions are one of the main reasons why free radicals can cause damage far from where they form. Any organ or system can be exposed to oxidative or nitrosative stress. However, the most susceptible to stress are the brain (high metabolic activity and low levels of endogenous antioxidants) and the circulatory system (fluctuations in oxygen and nitric oxide levels).
In AH, iNOS dysfunction is observed to a greater extent, since it is an isoform that catalyzes the formation of NO by endothelial cells. Li Q. et al. (2015) described the iNOS uncoupling mechanism. Tetrahydrobiopterin (BH4) is a key cofactor responsible for normal electron transfer from the reductase domain of one NOS2 monomer to the oxygenase domain of another monomer to form NO. When this cofactor is deficient, iNOS produces a superoxide anion instead of NO [18], which marks the induction of oxidative stress in the vessels. There are a number of studies that confirm this mechanism: with the use of an animal model [19][20] and in clinical trials [21][22][23].
Kelm M. et al. (1996) identified three causes of NO-dependent vasospasm (impaired NO-dependent vasodilation) in patients with AH: (1) reduced NO synthesis by endothelial cells due to impaired signal transduction and/or decreased NO synthase activity, (2) accelerated NO degradation in the vessel wall, and (3) structural disorders in the vessel wall leading to a general decrease in the dilator capacity of resistant arteries [12]. Endothelial cells release NO in response to shear stress and activation of various receptors. NO stimulates guanylyl cyclase to form 3′,5′-cyclic guanosine monophosphate, which leads to vasodilation of vascular smooth muscle cells. NO has a vasodilating and antiproliferative effect on smooth muscle cells and inhibits thrombocyte aggregation and leukocyte adhesion [11].
Increased NO biosynthesis enhances angiogenesis. Conversely, angiogenesis is impaired when NO levels are reduced (e.g., in NOS2 knockout animal models). That is, conditionally, angiogenesis begins in response to hypoxia. Thus, signs of impaired angiogenesis and, as a result, microvascular rarefaction are revealed in AH [24]. It is important that the degree of AH does not affect angiogenesis, and microvascular rarefaction is also observed in normotensive people with a family history of hypertension. The genetic basis of these mechanisms has also been studied. Recent studies have confirmed that the pathway of L-arginine conversion to NO is impaired not only in people with AH, but also in people with normotension with a history of essential AH [25].
Vascular tone is regulated by a variety of autocrine and paracrine systems. The vascular renin-angiotensin system, kallikrein-kinin system, natriuretic peptide system, endothelin, mechanosensitive ion channels, prostanoids, catecholamines, and endothelial hyperpolarizing factor are involved [12]. The impact on these systems is already actively used in AH therapy. Interestingly, the effects of many cardiovascular drugs used in hypertension affect the NO system. Activating NO signaling or increasing NO bioavailability are key mechanisms contributing to the positive cardiovascular effects of drugs [26]. Angiotensin-converting enzyme inhibitors, calcium channel blockers, third-generation beta blockers, and statins significantly improve endothelial function and NO bioactivity [27]. Angiogenic growth factors (vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF)) activate NOS because they require NO to function. They do this through effector molecules and their effect on the RAAS receptors. For example, bradykinin and angiotensin II induce angiogenesis [24].
Based on this, new treatment plans targeting the NO system are currently being investigated and developed; they include NO donorship and NOS stimulants [27]. In 1996, Preik M. et al. (1996) showed the effectiveness of NO donorship (glycerol trinitrate, isosorbide dinitrate and sodium nitroprusside), and also noticed that it depended not on the drug class, but on the severity of AH [28]. These data form the basis of modern research. Rajapakse N.W. et al. (2019) suggested that the decrease in NO bioavailability makes a significant contribution to the development of resistant AH. This means that NO donorship may be the most effective treatment in this cohort of patients [29]. Today, these are pilot projects and developments, and the first clinical trials. However, they are promising and of great scientific and clinical interest.
It is assumed that drug action on the NO system, in addition to other links of therapy, is especially advisable in the case of resistant AH and comorbidity with other diseases (heart failure, diabetes mellitus, obesity). NO-mediated endothelial dysfunction in such patients is more pronounced. Consequently, today, when developing drugs for AH treatment, special attention is paid to the NO system. For example, nebivolol (a third generation β-blocker) is highly selective for β1-adrenergic receptors and causes vasodilation through interaction with the L-arginine/NO endothelial pathway. Although nebivolol lowers blood pressure to the same extent as conventional β-blockers and other types of antihypertensive drugs, it will have a positive effect on the vascular endothelium [30]. Nebivolol releases NO, thereby preventing the development of hypertension associated with chronic NO deficiency, and this effect appears to be dependent on inhibition of the renin-angiotensin system [15].
3. Role of NO and NOSs in Pathogenesis and Treatment of TTH
TTH is a highly prevalent disorder with a significant impact on society. Understanding the pathophysiology of TTH is paramount for development of effective treatments and prevention of chronic TTH [31]. Advances in basic pain and clinical research have improved our understanding of the TTH pathophysiologic mechanisms [13]. Pain perception studies such as measurement of muscle tenderness, pain detection thresholds, pain tolerance thresholds, pain response to suprathreshold stimulation, temporal summation and diffuse noxious inhibitory control have played a central role in elucidating the pathophysiology of TTH [31]. Increased excitability of the central nervous system (CNS) generated by repetitive and sustained pericranial myofascial input may be responsible for the transformation of episodic TTH into the chronic form [13]. Molecular mechanisms that underlie TTH have not yet been clarified. Studies in which TTH was induced by intravenous infusions of glyceryl trinitrate (an exogenous NO donor) and histamine (which liberates NO from vascular endothelium) suggest that NO is likely to be a responsible molecule. The release of NO from blood vessels, perivascular nerve endings or from CNS tissue is an important molecular trigger mechanism in spontaneous headache pain [14]. Sarchielli P. et al. (2001) assessed the variations in L-arginine/NO pathway activity and platelet cyclic guanosine 3′,5′-monophosphate (cGMP) levels in patients affected by chronic TTH. A reduction in platelet aggregation response was found. The reduction in platelet aggregation was coupled with increased NO and cGMP production. A significant increase in cytosolic Ca(2+) concentration was also detected compared to control individuals. This was accompanied by a reduced platelet content and collagen-induced secretion of serotonin and increased content of NO in patients with TTH. The above findings were more pronounced in patients with analgesic abuse. It can be hypothesized that the increased NOS activity shown in platelets of TTH patients reflects an analogous central up-regulation of NOS activity in the spinal horn/trigeminal nucleus and supraspinal structures; these structures are involved in the modulation of nociceptive input from myofascial cranial structures contributing to central sensitization. The increase in NOS activity seems to be associated with a hyposerotonergic status, particularly in patients with analgesic abuse. This can contribute to central sensitization in patients with TTH. The increase in platelet glutamate content in the same patients suggests the implication of the above excitatory amino acid in spinal and supraspinal structures involved in head pain induction and maintenance [32].
Tenderness of pericranial myofascial tissues and number of myofascial trigger points are considerably increased in patients with TTH. Mechanisms responsible for the increased myofascial pain sensitivity have been studied extensively. Peripheral activation or sensitization of myofascial nociceptors could be one of the causes for increased pain sensitivity, but firm evidence for a peripheral abnormality is still lacking. Peripheral mechanisms are most likely of major importance in episodic TTH [33]. Sensitization of pain pathways in CNS due to prolonged nociceptive stimuli from pericranial myofascial tissues seems to be responsible for TTH chronification. Neck muscle nociception mediated by NO may play a role in TTH pathophysiology [16]. The role of NO in the antinociceptive effect of indomethacin was assessed in the pain-induced functional impairment model in the rat (PIFIR model); the antinociceptive effect of indomethacin involves, at least in part, the NO-cyclic GMP pathway at the peripheral level [34]. Furthermore, the role of NO in the antinoceptive effect of other drugs used for TTH treatment (diclofenac [35]; ketorolac [36]; nimesulide [37]; celecoxib [38]; rofecoxib [39]; gabapentin [40]; melatonin [41]) has been shown.
Chronic TTH may be caused by prolonged painful input from pericranial myofacial tissues, such as tender points, which results in central sensitization (increased excitability of neurons in the CNS). Animal studies have shown that sensitization of pain pathways may be caused by or associated with activation of nNOS and generation of NO. Furthermore, it has been shown that NOS inhibitors reduce central sensitization in animal models of persistent pain [42].
nNOS is involved in the induction but not the maintenance of nerve growth factor (NGF) caused by facilitation of nociception in the brainstem. The results from an experimental animal model may support the idea of nNOS and eNOS as potential targets for pharmacological treatment of TTH [16]. Infusion of α,β-methylene ATP (α,β-meATP) into murine neck muscle facilitates brainstem nociception. Unspecific NOSs inhibition prevents and reverses this sensitization. It is unclear whether nNOS, iNOS or eNOS isoenzymes are involved in this α,β-meATP effect. Ristic D. et al. (2010) provided evidence that nNOS plays a major role in induction and eNOS in maintenance of facilitation in neck muscle nociception. Divergent roles of NOS isoenzymes may promote research on target-specific treatments for headache and neck muscle pain [17].
The role of nNOS and iNOS in central sensitization induced by an intradermal capsaicin injection was investigated by Wu J. et al. (2001) [43]. Obtained results by Budziñski M. et al. (2000) suggest that NO derived from iNOS plays an inhibitory role in carrageenan-produced hyperalgesia in rat [44].
The analgesic effect of the NOS inhibitor L-N(G) methyl arginine hydrochloride was investigated. This drug significantly reduced headache and myofascial factors in patients with chronic TTH. These studies show that NO plays a crucial role in the pathophysiology of TTH. The analgesic effect of NOS inhibition in patients with chronic TTH is probably due to a reduction in central sensitization at the level of the spinal dorsal horn, trigeminal nucleus or both. Furthermore, NOS inhibition may become a novel principle in the future treatment of chronic TTH [42].
4. Role of SNVs of NOSs Genes in the AH and TTH Phenotype and Comorbidites
All three isoforms of the protein (nNOS, iNOS, eNOS) and its genes (NOS1, NOS2, NOS3) described above play key roles in the pathogenesis of both AH and TTH. They also negatively influence the leading environmental trigger of these nosologies: stress (Figure 2) and related neuropsychological disorders (anxiety and depression) [45].
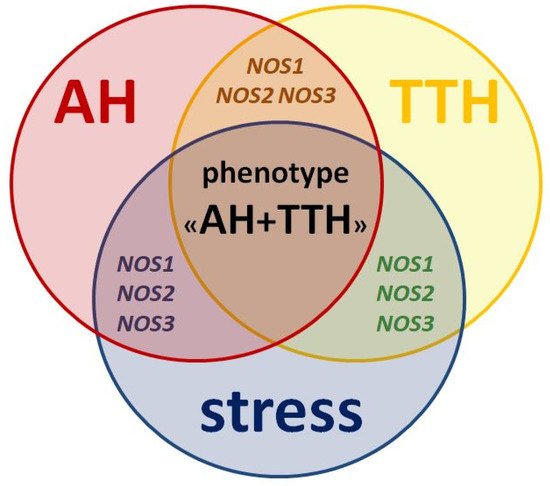
Figure 2. Similarities in pathogenesis and unresolved issues of the role of NOS1, NOS2, NOS3 genes as genetic predictors of the AH + TTH phenotype.
However, the prognostic role of NOS1, NOS2, NOS3 genes in the development of the common AH + TTH phenotype has not been studied in comparison with the second most common phenotype (AH + migraine). Our analysis testifies to the importance of planning and conducting associative genetic research in the role of NOS1, NOS2, NOS3 genes as genetic predictors of the AH + TTH phenotype (Figure 3) in various racial and ethnic groups.
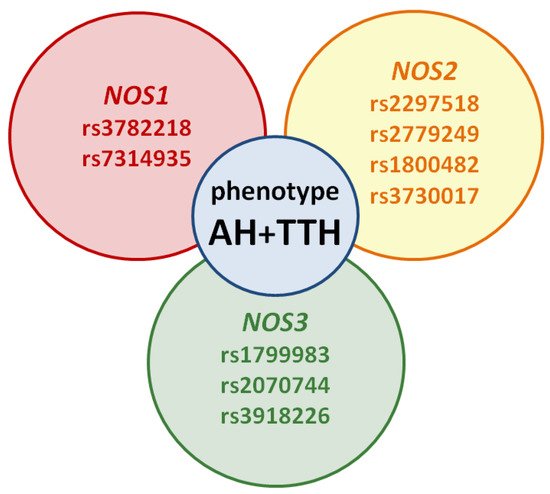
Figure 3. Potential SNVs of NOS1, NOS2, NOS3 genes predisposed to the AH + TTH phenotype.
This is important from a scientific and clinical point of view, because a new class of drugs that inhibit NOSs has been proposed in recent years, both for the treatment of AH and TTH. A new strategy for predicting and disease-modifying therapy of the common AH + TTH phenotype can increase the effectiveness and safety of treatment, improve patient’s quality of life, and reduce the risk of life-threatening cardiovascular complications.
References
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, J.E.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1∙25 million people. Lancet 2014, 383, 1899–1911.
- Chowdhury, D. Tension type headache. Ann. Indian Acad. Neurol. 2012, 15 (Suppl. S1), S83–S88.
- Petrova, M.M.; Moskaleva, P.V.; Shnayder, N.A.; Nasyrova, R.F. Comorbidity of arterial hypertension and tension-type headache (In Russ.). Kardiologiia 2020, 60, 132–140.
- Saccò, M.; Meschi, M.; Regolisti, G.; Detrenis, S.; Bianchi, L.; Bertorelli, M.; Pioli, S.; Magnano, A.; Spagnoli, F.; Giuri, P.G.; et al. The relationship between blood pressure and pain. J. Clin. Hypertens. 2013, 15, 600–605.
- Olesen, J. The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. Pharmacol. Ther. 2008, 120, 157–171.
- Ashina, M.; Bendtsen, L.; Jensen, R.; Olesen, J. Nitric oxide-induced headache in patients with chronic tension-type headache. Brain 2000, 123 Pt 9, 1830–1837.
- Mattila, J.T.; Thomas, A.C. Nitric oxide synthase: Non-canonical expression patterns. Front. Immunol. 2014, 5, 478.
- Hua, Y. Neuronal nitric oxide synthase in hypertension—An update. Clin. Hypertens. 2016, 22, 20.
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714.
- Hermann, M.; Flammer, A.; Lüscher, T.F. Nitric oxide in hypertension. J. Clin. Hypertens. 2006, 8 (Suppl. S4), 17–29.
- Kelm, M.; Preik, M.; Hafner, D.J.; Strauer, B.E. Evidence for a multifactorial process involved in the impaired flow response to nitric oxide in hypertensive patients with endothelial dysfunction. Hypertension 1996, 27 Pt 1, 346–353.
- Ashina, S.; Bendtsen, L.; Ashina, M. Pathophysiology of tension-type headache. Curr. Pain Headache Rep. 2005, 9, 415–422.
- Thomsen, L.L.; Olesen, J. Nitric oxide in primary headaches. Curr. Opin. Neurol. 2001, 14, 315–321.
- Fortepiani, L.A.; Ortíz, M.C.; Atucha, N.M.; García-Estañ, J. Nebivolol ameliorates nitric oxide deficient hypertension. Sci. World J. 2002, 2, 1676–1684.
- Isaak, A.; Ellrich, J. Neuronal nitric oxide synthase is involved in the induction of nerve growth factor-induced neck muscle nociception. Headache 2011, 51, 734–743.
- Ristic, D.; Spangenberg, P.; Ellrich, J. Inhibition of nNOS prevents and inhibition of iNOS reverses α,β-meATP-induced facilitation of neck muscle nociception in mice. Eur. J. Pharmacol. 2010, 647, 55–61.
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136.
- Shinozaki, K.; Nishio, Y.; Okamura, T.; Yoshida, Y.; Maegawa, H.; Kojima, H.; Masada, M.; Toda, N.; Kikkawa, R.; Kashiwagi, A. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ. Res. 2000, 87, 566–573.
- Podjarny, E.; Hasdan, G.; Bernheim, J.; Rashid, G.; Green, J.; Korzets, Z.; Bernheim, J. Effect of chronic tetrahydrobiopterin supplementation on blood pressure and proteinuria in 5/6 nephrectomized rats. Nephrol. Dial. Transplant. 2004, 19, 2223–2227.
- Ihlemann, N.; Rask-Madsen, C.; Perner, A.; Dominguez, H.; Hermann, T.; Køber, L.; Torp-Pedersen, C. Tetrahydrobiopterin restores endothelial dysfunction induced by an oral glucose challenge in healthy subjects. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H875–H882.
- Maier, W.; Cosentino, F.; Lütolf, R.B.; Fleisch, M.; Seiler, C.; Hess, O.M.; Meier, B.; Lüscher, T.F. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J. Cardiovasc. Pharmacol. 2000, 35, 173–178.
- Hambrecht, R.; Hilbrich, L.; Erbs, S.; Gielen, S.; Fiehn, E.; Schoene, N.; Schuler, G. Correction of endothelial dysfunction in chronic heart failure: Additional effects of exercise training and oral L-arginine supplementation. J. Am. Coll. Cardiol. 2000, 35, 706–713.
- Kiefer, F.N.; Neysari, S.; Humar, R.; Li, W.; Munk, V.C.; Battegay, E.J. Hypertension and angiogenesis. Curr. Pharm. Des. 2003, 9, 1733–1744.
- Schlaich, M.P.; Parnell, M.M.; Ahlers, B.A.; Finch, S.; Marshall, T.; Zhang, W.Z.; Kaye, D.M. Impaired L-arginine transport and endothelial function in hypertensive and genetically predisposed normotensive subjects. Circulation 2004, 110, 3680–3686.
- Pinheiro, L.C.; Tanus-Santos, J.E.; Castro, M.M. The potential of stimulating nitric oxide formation in the treatment of hypertension. Expert Opin. Ther. Targets 2017, 21, 543–556.
- Giles, T.D. Aspects of nitric oxide in health and disease: A focus on hypertension and cardiovascular disease. J. Clin. Hypertens. 2006, 8 (Suppl. S4), 2–16.
- Preik, M.; Kelm, M.; Feelisch, M.; Strauer, B.E. Impaired effectiveness of nitric oxide-donors in resistance arteries of patients with arterial hypertension. J. Hypertens. 1996, 14, 903–908.
- Rajapakse, N.W.; Giam, B.; Kuruppu, S.; Head, G.A.; Kaye, D.M. Impaired L-arginine-nitric oxide pathway contributes to the pathogenesis of resistant hypertension. Clin. Sci. 2019, 133, 2061–2067.
- Toblli, J.E.; DiGennaro, F.; Giani, J.F.; Dominici, F.P. Nebivolol: Impact on cardiac and endothelial function and clinical utility. Vasc. Health Risk Manag. 2012, 8, 151–160.
- Bezov, D.; Ashina, S.; Jensen, R.; Bendtsen, L. Pain perception studies in tension-type headache. Headache 2011, 51, 262–271.
- Sarchielli, P.; Alberti, A.; Floridi, A.; Gallai, V. L-Arginine/nitric oxide pathway in chronic tension-type headache: Relation with serotonin content and secretion and glutamate content. J. Neurol. Sci. 2002, 198, 9–15.
- Bendtsen, L.; Fernández-de-la-Peñas, C. The role of muscles in tension-type headache. Curr. Pain Headache Rep. 2011, 15, 451–458.
- Ventura-Martínez, R.; Déciga-Campos, M.; Díaz-Reval, M.I.; González-Trujano, M.E.; López-Muñoz, F.J. Peripheral involvement of the nitric oxide-cGMP pathway in the indomethacin-induced antinociception in rat. Eur. J. Pharmacol. 2004, 503, 43–48.
- Ortiz, M.I.; Torres-López, J.E.; Castañeda-Hernández, G.; Rosas, R.; Vidal-Cantú, G.C.; Granados-Soto, V. Role of the nitric oxide-cyclic GMP-K+ channel pathway in the antinociception induced by atrial natriuretic peptide (ANP) and diclofenac. Proc. West Pharmacol. Soc. 2002, 45, 174–177.
- Lázaro-Ibáñez, G.G.; Torres-López, J.E.; Granados-Soto, V. Participation of the nitric oxide-cyclic GMP-ATP-sensitive K+ channel pathway in the antinociceptive action of ketorolac. Eur. J. Pharmacol. 2001, 426, 39–44.
- Islas-Cadena, M.; Aguirre-Bañuelos, P.; Granados-Soto, V. Evidence for the participation of the nitric oxide-cyclic GMP pathway in the antinociceptive effect of nimesulide. J. Pharmacol. Toxicol. Methods 1999, 42, 87–92.
- Klein, T.; Eltze, M.; Grebe, T.; Hatzelmann, A.; Kömhoff, M. Celecoxib dilates guinea-pig coronaries and rat aortic rings and amplifies NO/cGMP signaling by PDE5 inhibition. Cardiovasc. Res. 2007, 75, 390–397.
- Déciga-Campos, M.; López-Muñoz, F.J. Participation of the NO-cyclic GMP pathway in rofecoxib-induced antinociception. Proc. West Pharmacol. Soc. 2003, 46, 165–167.
- Mixcoatl-Zecuatl, T.; Flores-Murrieta, F.J.; Granados-Soto, V. The nitric oxide-cyclic GMP-protein kinase G-K+ channel pathway participates in the antiallodynic effect of spinal gabapentin. Eur. J. Pharmacol. 2006, 531, 87–95.
- Hernández-Pacheco, A.; Araiza-Saldaña, C.I.; Granados-Soto, V.; Mixcoatl-Zecuatl, T. Possible participation of the nitric oxide-cyclic GMP-protein kinase G-K+ channels pathway in the peripheral antinociception of melatonin. Eur. J. Pharmacol. 2008, 596, 70–76.
- Ashina, M. Nitric oxide synthase inhibitors for the treatment of chronic tension-type headache. Expert Opin. Pharmacother. 2002, 3, 395–399.
- Wu, J.; Fang, L.; Lin, Q.; Willis, W.D. Nitric oxide synthase in spinal cord central sensitization following intradermal injection of capsaicin. Pain 2001, 94, 47–58.
- Budziñski, M.; Misterek, K.; Gumulka, W.; Dorociak, A. Inhibition of inducible nitric oxide synthase in persistent pain. Life Sci. 2000, 66, 301–305.
- Nasyrova, R.F.; Moskaleva, P.V.; Vaiman, E.E.; Shnayder, N.A.; Blatt, N.L.; Rizvanov, A.A. Genetic Factors of Nitric Oxide’s System in Psychoneurologic Disorders. Int. J. Mol. Sci. 2020, 21, 1604.


