1. Antioxidants—General Aspects and Main Determination Techniques
1.1. Defining, Classifying and Describing Modes of Action Antioxidants
Antioxidants are chemical species that prevent or delay oxidation processes. They originate from various sources and hamper lipid peroxidation following different mechanisms of intervention, acting at a much smaller concentration, in comparison to that of the preserved compound [1][2][3][4][5].
Primary antioxidants act as scavenging or chain breaking antioxidants, delaying initiation or disrupting propagation. Secondary antioxidants quench singlet oxygen, decompose peroxides in non-radical species, chelate prooxidative metal ions, inhibit oxidative enzymes or absorb UV radiation. It has been confirmed that they can exploit the above-described mechanisms to stabilize/regenerate primary antioxidants [1].
Considering antioxidants’ reactivity, four lines of defense have been described. Antioxidants belonging to the first line of defense withhold radical species generation. The second line of defense includes mainly radical scavenging antioxidants. The third line of defense intervenes after the free radical-caused insults, being composed of repair antioxidants. Adaptation mechanisms underlie the mode of action of the fourth line of defense: Signals required for free radical generation are exploited, thus such antioxidants can disrupt free radical occurrence or reactions implying radical species intervention [6][7].
Superoxide dismutase, catalase, glutathione peroxidase, metal-binding proteins like lactoferrin, ferritin, caeruloplasmin, glutathione, uric acid, alpha-lipoic acid, ubiquinones, bilirubin, and melatonin are well-recognized endogenous antioxidants. Tocopherols, phenolics, vitamin C, and carotenoids are exogenous antioxidants found in food and /or dietary supplements, slowing up the use of endogenous antioxidants, so the cell’s own antioxidant profile can remain unaltered [1][8].
Synthetic antioxidants such gallic acid esters, synergistic butylated hydroxyanisole and butylated hydroxytoluene are added to foodstuffs to prevent rancidity. Another antioxidant classification takes account on the solubility: Hydrophilic (ascorbic acid, glutathione, uric acid, flavonoids) and lipophilic (carotenoids, tocopherols, ascorbyl palmitate, or stearate) antioxidants [1].
With respect to the mechanism involved in free radical inactivation, antioxidant can follow either hydrogen atom transfer, or single electron transfer. Hydrogen atom transfer is swift and is not dependent on pH or nature of solvent, but proved sensitive to the presence of other reductant species. The behavior of an antioxidant molecule can also encompass single electron transfer [9][10]. Considering the analytical methods developed, hydrogen atom transfer underlies Oxygen Radical Absorbance Capacity (ORAC), Total Radical Trapping Antioxidant Potential (TRAP) and chemiluminescence, whereas single electron transfer underlies Ferric reducing ability of plasma (FRAP), and Cupric Reducing Antioxidant Capacity (CUPRAC) [2][10]. 2,2-diphenyl-1-picrylhydrazyl (DPPH) and Trolox Equivalent Antioxidant Capacity (TEAC) can exploit both mechanisms [10][11].
Antioxidants can hamper the deleterious effects of free radicals in the human body, as well as the oxidative decay of food components [1][2][3][12][13][14]. Although both terms have been employed in papers approaching antioxidant assay, distinction has been drawn between antioxidant activity and antioxidant capacity. The antioxidant capacity reflects the conversion of the reactive oxygenated species that is scavenged. This illustrates the scavenging ability, and can be expressed as the amount, as moles, of the scavenged free radical by antioxidants [15][16], present in an analyzed sample, for instance a plant extract [17]. The term “antioxidant activity” is prevalent in electrochemical approaches, that directly provide informations about analyte concentration. Hence, “antioxidant activity” is linked to a thermodynamic significance, as it can be correlated to the total active or effective concentration of antioxidants, or oxidants in a sample. It can be expressed as units of standard antioxidant, for instance milligrams, mmoles or μmoles Trolox equivalents or other reference antioxidant (ascorbic acid, gallic acid, quercetin, catechin, rutin), per amount (grams, kilograms, liters, etc.) of sample. “Antioxidant power” and “antioxidant ability” are less used, and they do not have a precise interpretation [18].
1.2. Analytical Methods Applied to Antioxidant Determination
1.2.1. General Overview of Methods
Antioxidant assay can rely on a plethora of methods, based on electrochemical, spectrometrical or chromatographic detection [4][19][20]. A synoptic overview of the principles underlying the main analytical techniques and detection systems applied to antioxidant assay is presented in [21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62].
Table 1. The main analytical methods applied to antioxidant assay [21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62].
| Total Antioxidant Capacity or Its Main Contributors’ Assay |
Method’s Principle |
Detection of the End-Product |
Ref. |
| |
Spectrometry |
|
|
| DPPH |
Antioxidants react with an organic radical |
Colorimetric |
[21][22] |
| ABTS |
Antioxidants react with an organic cation radical |
Colorimetric |
[23][24] |
| FRAP |
Antioxidants react with ferric-tripyridyltriazine complex |
Colorimetric |
[25][26] |
| PFRAP |
Potassium ferricyanide is reduced by antioxidants to potassium ferrocyanide, that reacts with Fe3+ yielding ferric ferrocyanide |
Colorimetric |
[27][28] |
| CUPRAC |
Antioxidants reduce Cu (II) complex to a Cu (I) complex |
Colorimetric |
[29][30] |
| Thiobarbituric Acid Reactive Species (TBARS) Assay |
The generation of malonyl dialdehyde can be detected after its reaction with thiobarbituric acid, yielding a pink chromogen |
Colorimetric assay of malonyl dialdehyde-thiobarbituric acid adduct |
[31][32] |
| Folin-Ciocalteu |
Phenolics react with a mixture of phosphomolybdate/phosphotungstate in the presence of sodium carbonate 20%. |
Absorbance of the blue molybdenum-tungsten complex resulted is measured, versus gallic acid as reference antioxidant |
[33][34] |
| ORAC |
Peroxyl radicals, induced by AAPH
(2,2′-azobis-2-amidino-propane) decomposition are reduced by antioxidants |
Loss of fluorescence indicated by fluorescein |
[35][36] |
| HORAC |
Co(II)-based Fenton systems result in OH radicals generation, followed by quenching by antioxidants |
Loss of fluorescence indicated by fluorescein |
[37][38] |
| TRAP |
Luminol-derived radicals, formed by AAPH decomposition, are scavenged by antioxidants |
Quenching of chemiluminescence |
[39][40] |
| Fluorimetry |
Emission of electromagnetic radiation (generally in the visible range) that follows an absorbtion process (generally in the UV domain) |
Recording of excitation/emission spectra of fluorescent reagent |
[41][42] |
| |
Electrochemical Techniques |
|
|
| Potentiometry |
Antioxidants interact with the oxidized form of a redox couple, changing the ratio between the oxidized form and the reduced form concentration |
The analytical signal recorded is the potential shift of the mediator system, resulting from interaction with antioxidants |
[43][44] |
| Cyclic voltammetry (CV) |
Linear variation of the potential of a working electrode following a triangular waveform, and recording of current intensity |
The intensity value corresponding the cathodic/anodic peak is measured |
[45][46] |
| Differential pulse voltammetry (DPV) |
Voltage pulses are superimposed on the potential scan, that is varied linearly or stairstep-wise |
First intensity value is sampled before applying the pulse, and the second towards the end of the pulse |
[47][48] |
| Square-wave voltammetry (SWV) |
A square wave is superimposed on a potential staircase sweep variation |
Current intensity recorded at the end of each potential change |
[49][50] |
| Polarography |
Determination of the antioxidant potential of radical scavengers relied on the anodic oxidation of dropping mercury electrode |
Diminution of the anodic limiting current of the hydroxoperhydroxo-mercury(II) complex, [Hg(O2H) (OH)], generated in H2O2 solution at alkaline pH, at the potential of Hg oxidation |
[51][52] |
| Amperometry |
Measurement of the current intensity at a fixed potential value of the working electrode, with respect to a reference one |
The intensity of the current, occurring as result of oxidation/reduction of the analyte at constant potential, is measured |
[53][54] |
| Biamperometry |
Reaction of the antioxidant with the oxidized form of a reversible indicator redox couple |
The current flowing between two identical working electrodes is measured, at a small potential difference; the measuring solution contains the antioxidant(s) in the presence of a reversible redox couple |
[55][56] |
| |
Chromatography |
|
|
Gas chromatography
(GC) |
The compounds to be separated and quantified are differentially distributed between a liquid stationary phase and a gaseous mobile phase |
Detection based on thermal conductivity or flame ionisation |
[57][58] |
High performance liquid chromatography
(HPLC) |
The compounds to be separated suffer different repartition between a solid stationary phase and a liquid mobile phase with various polarities, at high values of pressure of the mobile phase and flow rate |
Diode array (UV-VIS), mass spectrometry, fluorescence, or electrochemical detection |
[59][60] |
Thin layer chromatography
(TLC) |
Compound separation relies on the repartition between a solid stationary phase (silica gel, alumina) and a liquid mobile phase (methyl acetate/formic acid, ethanol/hexane, or methanol/chloroform/ hexane) |
UV-VIS vizualization, fluorescence or phosphorescence detection |
[61][62] |
2. Carbon Electrodes—General Overview
Carbon electrodes are largely employed in electroanalysis due to their special features, that encompass large surface area, tunable porosity, high electroconductivity, chemical stability, temperature resistance, nanostructuring possibilities, facility of manufacturing at low cost, and easiness of surface modification. The carbonaceous electrodes were classified as carbon paste, glassy carbon, fullerenes, graphite, diamond, and screen- printed electrodes [63].
Carbon Paste Electrodes: Carbon paste electrodes are synthesized from graphite powder and various water-immiscible nonelectrolytic organic pasting liquids, such as mineral (paraffin) oil, [64][65]. Most often high purity mineral oil (Nujol) is employed, nevertheless quasi-solid binders such as silicone grease or polypropylene could replace commonly used pasting liquids, but the developed structure has much higher density and becomes less easy to handle [66]. The advantages of carbon paste electrodes are facility of including modifiers (for developing novel, redox-mediated sensors), very low ohmic resistance, minimized toxicity of this environmentally compatible material, reduced background current, individual polarizability [63]. Obtaining carbon paste electrodes can also be rely on alternative carbon-based materials, replacing graphite powder with glassy carbon powder, carbon nanotubes, porous carbon foam, acetylene black [66].
Glassy Carbon Electrodes: Glassy carbon, also called vitreous carbon is a type of nongraphitizing, solid, three-dimensional carbon material, broadly used in electro-assay. It is obtained at temperatures above 2000 °C, to decompose pyrolysis intermediates that generally exhibit a scarce thermal conductivity [67]. The surface of glassy carbon electrodes can be modified with functional nanomaterials (metals, alloys, or metal oxides) [68][69]. Glassy carbon and glassy carbon-based electrodes provide excellent electroconductivity, mechanical resistance, broad potential range, and gas impermeability [63]. They have large potential window and chemical stability, prove better resistance to solvents than metal electrodes, been confirmed for their viability at the assay of organic compounds. Bare glassy carbon electrodes are characterized by facility of use, being mechanically cleaned by mere polishing on alumina slurry, procedure that can be followed by sonication in aqueous medium. Nevertheless, residual alumina particles on the electrode surface can affect the recorded electrochemical profile of electroactive analytes endowed with reductive (antioxidant) potential, such as phenolics [70].
Glassy carbon modification with alumina particles aimed at improving sensitivity in the case of dopamine [71]. The application of alumina-modified glassy carbon electrode promotes sensitivity, detectability and selectivity in the case of nitroaromatic compounds. Enhancement of dissolved oxygen electrochemical reduction in the presence of alumina was also reported [72]. Employing alumina suspensions for modifying glassy carbon electrodes by abrasive polishing, resulted in successful electro-assay of various phenolics. Modification of glassy carbon electrodes with α-alumina resulted in improved electrochemical response in comparison with θ and γ–alumina, and it was confirmed that alumina structure, and not the particle size or surface area, can exert notable effects [73].
Glassy carbon modification with alumina enhanced the voltammetric current response of gallic acid, caffeic acid, chlorogenic acid, catechin, quecetin, and rutin. By applying this simple procedure of electrode modification, lowered DPV relative standard deviation (RSD < 3%, n = 5) for repetitive assays and high inter-electrode precision (RSD < 4%, n = 3) were reported. Nevertheless, when use of unmodified glassy carbon electrodes is chosen, the application of the sonication step to remove all residual alumina is compulsory, as alumina remaining on the electrode surface can significantly affect the voltammetric profile of sample antioxidants [74].
The use of other metal oxides for modification, may also provide performance improvement in the electrochemical determination of antioxidant species. Modification with metal (gold, silver and platinum) or metal oxide nanoparticles imparts distinctive size-dependent electrochemical features. The sol-gel chemistry served for developing silica-modified electrodes, exploiting silica adsorption properties [74][75].
Carbon nanomaterials were divided into: Zero-dimensional fullerenes [76], one dimensional carbon nanotubes [77], two-dimensional graphene [78], and the three-dimensional porous carbons [79]. Porous carbon materials are fabricated using precursors named template composite materials which are synthesized, with subsequent carbonization and template removal [80]. Nevertheless, such a technique is laborious and necessitates a series of synthetic steps, the first stage of template injection, the etching process and the long solidification time [81], which may restrict mass applications of porous carbon materials [82].
Fullerene Electrodes: Fullerenes represent a class of carbon compounds in which carbon atoms form closed cage or cylinder-shaped structures. In the first case the compound is called Buckminsterfullerene (C60, named after the American architect R. Buckminster Fuller, whose geodesic dome was constructed relying on the same structural principles), and in the second case, the obtained structure is called carbon-nanotube [83][84][85]. Single-walled and multi-walled carbon nanotubes are widely employed in electrode modification, being biocompatible, having large surface area to volume ratio, enhanced electro-conductivity, chemical and mechanical resistance.
In the structure of graphene, atoms constitute a single layer and are placed in a two-dimensional honeycomb lattice. Each atom uses sp2 hybridized orbitals to connect by sigma bonds to its three nearest neighbors, and contributes with one electron (belonging to the p unhybridized orbital) to the conduction band that is common for the whole sheet. This type of bond is also encountered in carbon nanotubes, fullerenes and glassy carbon. Graphene oxide nanoparticles, alongside metal oxide nanoparticles are largely used in electrode modification, aiming at antioxidant determination. The oxygenated groups can lead to improvement in the electrochemical responses and mechanical properties. The polarity of these groups present at the surface of graphene oxide results in high dispersibility in polar solvents, enabling applications in biosensing systems.
Graphite Electrodes: Graphite is an allotropic form of carbon, where sp2-hybridized atoms form planes of hexagonal bonds. Graphite is a stable crystalline form of carbon, which can be employed as such, or in the form of composites [86][87][88][89]. Electrodes are inexpensive, commercially available and easy to modify, so they benefit from selectivity enhancement through various modifications, renewable surfaces and hazard-free polishing [90]. The high delocalization degree of pi electrons and the weak van der Waals interactions between the layers, result in good electro-conductivity.
Diamond Electrodes: Diamond is an allotrope form of carbon with insulating properties, very good mechanical resistance, the hardness being due to the bonds established between sp3 carbon atoms. Diamond is a non-easily accessible material, whose preparation require high temperatures and pressures [91]. By doping diamond with boron in different proportions, conductive, superconducting or semiconductor materials can be synthesized. These electrodes are chemically inert and are endowed with excellent electrical features. High boron-doped (103–104 ppm) diamond has metal-like conductivity and can be applied as electrode material [92]. Boron-doped polycrystalline diamond exhibits a rougher morphology, a higher sp3 content, a broader water potential window, and a lower background current [93].
Screen-Printed Electrodes: Such electrodes are developed by printing inks on ceramic or plastic surfaces. The inks, depending on the composition (carbon, gold, platinum) will determine the characteristics of screen-printed electrodes, that can be part of a three electrode set-up in a measuring cell. Depending on the target analyte, the ink can be modified by incorporation of metal powders, redox complexes or biocatalysts, to promote electron transfer [94][95][96].
Glassy carbon, vitreous carbon, carbon nanotubes, fullerenes, screen-printed, graphite, and diamond electrodes are considered homogenous carbon electrodes, whereas carbon paste and modified carbon pastes are heterogeneous carbon electrodes.
In a recent study, a detailed description of advantages and shortcomings of several broadly employed types of carbon-based electrodes is provided [97].
Carbon paste electrodes offer an analytical response on wide potential ranges, with small background current, reduced ohmic resistance, and facility of tuning pretreatment methods and surface modification. These materials do not harm the environment and operate at low cost. Nevertheless, they were characterized as not stable for functioning in flow systems, are not compatible with organic solvents, and often require recalibration. When using organic compounds as binders, the use of such sensors results in irreversibility of recorded voltammograms, and the surface roughness can influence reproducibility of the response.
Glassy carbon electrodes exhibit high mechanical resistance and excellent electro-conductivity. They are characterized by chemical inertness and function on large potential domains, over wide pH ranges, from strongly acidic to alkaline environment. Their large size and difficulty to manufacture at large scale may constitute inconvenients. The electron transfer occurs slower than at electrodes based on noble metals.
Carbon fiber microdisk electrodes are easy to use due to their small diameter and benefit from high speed of electron transfer. They give a rapid analytical response and exhibit high sensitivity for small concentration changes. Being compatible with biological media and non-toxic to cells, can be applied successfully to in vivo determinations. Nevertheless, they may not exhibit resistance to the mechanical force or to the high temperatures applied during the step of capillary pulling. Low selectivity and the possibility to break glass insulation during in vivo measurements are other disadvantages.
Basal plane pyrolytic graphite electrodes are characterized by high speed electrode reaction kinetics, with lowered background signal. Shortcomings may be the irreversible behavior and the large dimensions.
Screen-printed carbon electrodes are easy to employ, due to portability and facility to apply modifications. A broad series of geometries can be obtained, and the determinations can be performed with the possibility to eliminate surface fouling, and low cost. Nevertheless, the incorporated binders may alter the shape of voltammograms. Fouling of the electrode surface may be caused by products of redox reactions. Other shortcomings may be the rough surface and the slow reaction kinetics. The organic solvents present in the buffer may dissolve the ink, diminishing sensitivity.
Given the confirmed advantages of boron-doped diamond (excellent electroconductivity, mechanical resistance), the behavior of this type of electrode has been investigated in cyclic voltammetry of gallic acid. At low potentials, when the electrolytes are stable, deactivation of boron-doped diamond has been reported. It was found that gallic acid electro-oxidation generated the occurrence of a polymeric film on the anodic surface, causing boron-doped diamond deactivation [98].
3. Determination of Individual Antioxidants with Carbon-Based Electrodes
The determination of individual key antioxidants relied on a series of unmodified or modified carbon-based electrodes. An overview of the analytical parameters and applications on real samples, at the assay of some individual antioxidants, using carbonaceous working electrodes is presented in [99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143].
Table 2. Electroanalytical techniques applied to the assay of key individual antioxidants [99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143].
| No |
Antioxidant |
Analytical Method |
Carbon-Based Working Electrode |
Analytical Characteristics |
Ref. |
| 1. |
Ascorbic acid (AA) |
Cyclic
voltammetry;
Differential pulse
voltammetry; |
-carbon paste electrode; |
-linear range 0.07–20 mM
-supporting electrolyte: KCl 0.1 M;
-RSD 2.35% in DPV and 2.29% in CV;
-LOD 0.018 mM (CV) and 0.02 mM (DPV), calculated as 3× square mean error (for 10 determinations of the blank)/the slope of the calibration graph;
-LOQ 0.062 mM (CV) and 0.068 mM (DPV), calculated as 10× square mean error (for 10 determinations of the blank)/the slope of the calibration graph;
-analysis of commercial and home-made fruit juices;
-oxidation peaks at 470 mV in DPV and at 510 mV in CV (vs SCE); |
[99] |
| 2. |
Ascorbic acid |
Differential pulse
voltammetry; |
-screen-printed carbon electrode; |
-determinations performed in phosphate buffer solutions (pH: 5.8, 7.0 and 7.4);
-oxidation peak potentials increasing with concentration, noticed between −0.02 V and 0.11 V (vs. Ag/AgCl);
-linear range of 1 to 4 µM (pH 5.8) and 2–10 µM (pH 7.0), as present in calibration curve;
-assay of injectable vitamin C solutions; |
[100] |
| 3. |
Ascorbic acid
Dopamine
Paracetamol |
Cyclic voltammetry;
Differential pulse voltammery;
Chronoamperometry; |
-platinum nanoparticles-decorated graphene nanocomposite electrode, compared with graphene-modified glassy carbon electrode and bare glassy carbon electrode; |
-supporting electrolyte: KCl 0.1 M;
-linear range 300 μM to 20.89 mM (for AA in CV);
-LOD of 300 μM (for AA in CV);
-AA exhibited two linear DPV ranges, 300 μM to 7.36 mM and 8.12 to 39.87 mM;
-LOD 5 μM (AA in DPV);
-linear range from 420 μM to 29.26 mM for ascorbic acid in chronoamperometry, at 0.0 V vs. Ag/AgCl used as reference; |
[101] |
| 4. |
Ascorbic acid |
Cyclic voltammetry;
Chrono-amperometry; Linear sweep voltammetry (LSV); |
-carbon veil electrode modified with phytosynthesized gold nanoparticles; |
-LSVs recorded from 0.0 V to +0.8 V, vs. Ag/AgCl, at a scan rate of 0.05 Vs−1;
-supporting electrolyte: Phosphate buffer pH 5.0 to 8.0;
-modification with gold nanoparticles shifted the cyclic voltammetric potential of AA oxidation with more than 0.4 V towards less positive values;
-linear response to AA 1 µM–5.75 mM in anodic voltammetry;
-LOD 0.05 µM and LOQ 0.15 µM (CV);
-most increased oxidation current of AA obtained in pH 6.0 phosphate buffer solution;
-analysis of fruit juices; |
[102] |
| 5. |
Ascorbic acid
Uric acid
Cholesterol |
Cyclic voltammetry;
Square-wave voltammetry; |
-carbon paste electrode modified with copper oxide-decorated reduced graphene; |
-supporting electrolyte: Phosphate buffer pH 7.4;
-scan rates (CV) of 10, 25, 50, 100, 150, 200, 250, 300, 500 mV s−1, for 500.0 µM cholesterol, using Ag/AgCl as reference;
-linear response to AA 0.04–240.0 µM, with a LOD of 9 nM (SWV);
-linear response to uric acid 0.04–400 µM, with a LOD of 8 nM (SWV);
-linear response to cholesterol 0.03–300 µM, with a LOD of 9 nM (SWV);
-differences between peak potentials (SWV) as follows: 430 mV (between cholesterol and ascorbic acid), 270 mV (between ascorbic acid and uric acid) and 700 mV (between cholesterol and uric acid); |
[103] |
| 6. |
Delphinidin, Cyanidin, Pelargonidin, Kuromanin, Callistephin |
Cyclic voltammetry;
Differential pulse voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Methanol containing 0.1 mol L−1 lithium perchlorate or 0.1 mmol L−1 Britton-Robinson buffer;
-pulse amplitude of 50 mV, pulse width of 70 ms, and scan rate of 10 mV s−1 (DPV);
-cyclic voltammetric scan rates ranging from 25 mV s−1 to 500 mV s−1;
-oxidation peak potentials comprised between 519 and 1115 mV vs Ag/AgCl (CV);
-the larger the number of hydroxyl groups in the B ring, the lower the oxidation potential;
-sugar moieties result in displacement of peak potentials to more positive values; |
[104] |
| 7. |
Oenin chloride;
Malvin chloride;
Kuromanine chloride;
Cyanin chloride;
Myrtillin chloride;
Petunidin chloride; |
Cyclic voltammetry;
Differential pulse voltammetry;
Square-wave voltammetry; |
-glassy carbon electrode; |
-supporting electrolytes: Acetic acid/acetate buffer pH 3.5 and 4.5, as well as phosphate buffer pH = 7.0;
-voltammetric scans in the potential range of 0 to + 1.4 V vs. Ag/AgCl;
-differential pulse voltammetric pulse amplitude 50 mV, pulse width 70 ms and scan rate 5 m Vs.−1;
-square-wave voltammetric frequency 13, 25 and 50 Hz; amplitude 50 mV and potential increment 2 mV;
-first cyclic voltammetric oxidation peak appears at 0.3 V for kuromanine chloride as well as for cyanin chloride, with a corresponding cathodic peak at 0.23 V in phosphate buffer pH = 7.0;
-kuromanine chloride showed a DPV oxidation peak potential at 0.49 V and peonidin-3-O-glucoside at 0.39 V, in 0.2 M acetate buffer, pH 3.5; |
[105] |
| 8. |
Delphinidin-3-O-glucoside;
Malvidin-3-O-glucoside-catechin;
Peonidin-3-O-glucoside-4-vinylphenol, etc.; |
Cyclic voltammetry;
Differential pulse voltammetry; |
-glassy carbon electrode; |
-supporting electrolytes: Acetate-acetic acid buffer pH 3.6, and acetate-acetic acid buffer pH 3.6, containing 12% ethanol;
-cyclic voltammograms obtained in the range of 0 to +0.8 mV, at a scan rate of 100 mV/s;
-differential pulse voltammetric measurements performed with a pulse amplitude of 50 mV and a pulse width of 50 ms;
-20-fold diluted wine presented DPV peak potentials of 443 mV and 666 mV vs Ag/AgCl, similar to those of wine extract, and to those of malvidin-3-O-glucoside (53.6% of the total anthocyanin content in grape extract);
-20-fold diluted wine presented one oxidation peak, at 491 mV in CV;
-ascorbic acid (0.05–0.1 mg/mL) used as reference, presented an oxidation peak at 270 mV in DPV; |
[106] |
| 9. |
Malvidin-3-glucoside, Catechin, Epicatechin, Gallic acid, Hydroxycinnamic acids, etc. |
Square-wave voltammetry; |
-glassy carbon electrode; |
-disposable unmodified screen-printed carbon electrodes;
-screen-printed carbon electrodes modified with single- and multi-walled carbon nanotubes;
-determinations performed in a model wine solution: 12% (v/v) ethanol, containing 33 mM l-tartaric acid, at pH 3.6;
-Ag/AgCl as reference electrode;
-characterization of red wine polyphenols;
-at the single-walled carbon nanotubes-modified screen-printed carbon electrode, first peak obtained between 138 mV (gallic acid) and 340 mV (malvidin-3-O-glucoside);
-at the multi-walled carbon nanotubes-modified screen-printed carbon electrode, first peak obtained between 120 mV (gallic acid) and 370 mV (malvidin-3-O-glucoside); |
[107] |
| 10. |
β-carotene |
Cyclic voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: 0.1 M LiClO4 in ethanol containing 10% CH2Cl2;
-potential scan rate of 100 mV s1;
-potential range from 0 to 1500 mV;
-beta-carotene irreversibly oxidized at 500 and 920 mV vs Ag/AgCl reference;
-linear analytical range of 10 to 380 mM;
-LOD 2.5 mM;
-LOQ 8.3 mM; |
[108] |
| 11. |
β-caro
tene |
Square-wave voltammetry; |
-paraffin impregnated graphite electrode; |
-supporting electrolyte: 0.1 M HClO4;
-pulse amplitude 50 mV;
-step potential 2 mV;
-SWV showed oxidation peaks at 0.88 V and 1.09 V versus Ag/AgCl for β-carotene and astaxanthin;
-analysis of raw vegetables and fruits; |
[109] |
| 12. |
β-carotene |
Chrono-amperometry; |
-stochastic sensor based on a graphene–porphyrin composite; |
-supporting electrolyte: Acetate buffer pH = 3.0
-working potential of 125 mV versus Ag/AgCl;
-linear response in the range between 1.0 × 10−15 mol L−1 and 1.0 × 10−3 mol L−1;
-LOQ 1.0 × 10−15 mol L−1;
-sensitivity 8.66 × 1010 s−1/mol L−1;
-analysis of soft drinks; |
[110] |
| 13. |
Astaxanthin |
Square-wave voltammetry; |
-paraffin-impregnated graphite rod electrode |
-two electrolyte solutions: 0.1 mol L−1 HClO4 and 0.1 mol L−1 KNO3;
-frequency of 100 Hz, pulse amplitude 50 mV and step potential 2 mV.
-LOD 15.77 µmol L−1 and LOQ 47.80 µmol L−1;
-first reversible oxidation at −0.276 V vs. Ag/AgCl; second, not well defined, oxidation peak at −0.032 V; third reversible voltammetric response at 0.335 V; |
[111] |
| 14 |
Quercetin, dihydroquercetin, ferulic acid, synapic acid, gallic acid, caffeic acid etc. |
Cyclic voltammetry; |
-pyrographite electrode; |
-determinations performed in: 0.2 M potassium phosphate buffer pH 6.0; 0.05 M potassium citrate buffer pH 5.0, and 0.1 M citrate-phosphate buffer pH 3.5; for increasing conductivity, 0.1 M KCl was added as an auxiliary electrolyte;
-scanning speed 25 mV/s;
-analyzed phenolics showed oxidation peaks in the range 235–834 mV, vs. Ag/AgCl; |
[112] |
| 15. |
Catechin, caffeic acid, coumaric acid, syringic acid, quercetin, mailvidin trans-resveratrol;
estimation of total polyphenols levels; |
Cyclic voltammetry; |
-glassy carbon electrode |
-determinations performed in model wine solution, consisting of 12% (v/v) ethanol, 33 mM l-tartaric acid, pH = 3.0, with Ag/AgCl as reference;
-the anodic peak area in the range −100 to 1200 mV accounted for about 70% of total phenolics that absorbed at 280 nm;
-catechol and galloyl containing polyphenols present in wine were quantitated relying on the size of the first anodic peak at around 450 mV after treatment with acetaldehyde;
-flavonols were quantitated on the basis of the anodic peak current at 1120 mV;
-good correlation of total flavanols with HPLC; |
[113] |
| 16. |
Catechin;
estimation of total polyphenols levels; |
Differential pulse
voltammetry; |
-glassy carbon electrode modified with green apple-sourced polyphenol oxidase (biosensor); |
-supporting electrolyte phosphate buffer pH 7.65;
-the anodic peak for reversible catechin oxidation, noticed at 0.219 V, with a cathodic peak at 0.128 V vs Ag/AgCl reference;
-LOD 1.76 μg L−1;
-LOQ 5.86 μg L−1;
-RSD 2.5%;
-detection of polyphenols in wine; |
[114] |
| 17 |
α-tocopherol; |
Square-wave voltammetry; |
-carbon fiber disk ultramicroelectrode |
-determinations performed in benzene/ethanol and 0.1 mol L−1 H2SO4;
-square wave amplitude 50 mV;
-staircase step height 0.005 V;
-frequency 25 Hz;
-peak potential between 0.6 and 0.7 V versus SCE; |
[115] |
| 18. |
α-tocopherol; |
Cyclic voltammetry; |
-glassy carbon electrode |
-supporting electrolyte: Glacial acetic acid and acetonitrile, containing 0.4 M sodium perchlorate;
-100 mV s−1 scan rate;
-external silver chloride reference;
-peak potential 548 mV at first electron loss (that leads to phenoxyl radical) and 517 mV for second electron loss (that leads to phenoxonium cation radical); |
[116] |
| 19. |
α-tocopherol; Gallic acid; ascorbic acid; |
Cyclic voltammetry; |
-hydrophilic indium tin oxide electrode, lipophilic fluorinated nanocarbon film electrode and glassy carbon electrode; |
-supporting electrolyte: Phosphate buffer saline (pH = 7.0), sodium dodecyl sulfate surfactant, 2-butanol cosurfactant, and toluene;
-antioxidants analyzed in bicontinuous microemulsion, in which water and oil phases coexisted at microscopic scale;
-using the indium tin oxide electrode, hydrosoluble gallic acid, ascorbic acid, and amphiphilic trolox exhibited irreversible anodic oxidation peaks at 0.61, 0.41, and 0.72 V, respectively, vs saturated calomel;
-using the lipophilic fluorinated nanocarbon film electrode, amphiphilic trolox and lipophilic α-tocopherol, gave irreversible oxidations at 0.90 and 0.69 V, respectively, vs saturated calomel; |
[117] |
| 20. |
α-tocopherol; |
Square wave anodic stripping voltammetry; |
-glassy carbon paste electrode, |
-supporting electrolyte: 0.1 M HNO3;
-linear ranges of 5 × 10−7− 4 × 10−5 and 5 × 10−8− 1×10−5 mol L−1;
-LOD of 1 × 10−7 mol L−1;
-anodic peak potential at 520 mV vs. Ag/AgCl reference,
-analyte extracted into glassy carbon paste electrode with 10% silicone oil, from 60% aqueous-acetonic mixture;
-analysis of margarine and edible oils; |
[118] |
| 21. |
Superoxide dismutase; |
Amperometry; |
-glassy carbon electrode; |
-detection performed at 0.0 V vs saturated calomel reference
-LOD 8 × l0−11 M at pH 7.0, and 2 × 10−12 M at pH 9.0;
-assay of buttermilk-sourced superoxide dismutase solution, in 0.1 M phosphate buffer, pH 8.0, containing 1 × 10−4 M EDTA; |
[119] |
| 22. |
Superoxide dismutase; |
Cyclic voltammetry; |
-screen-printed carbon electrode modified with self-assembled monolayers of gold nanoparticles in electropolymerized polypyrrole, and biofunctionalized with monoclonal anti-SOD1 antibody (immunosensor); |
-supporting electrolyte: 0.1 M phosphate buffer solution containing 100 μM nitrite;
-scan rate of 50 m Vs−1, using Ag/AgCl reference;
-peak current recorded at the potential 0.8 V for UV-A treated cells was significantly higher than for control cells;
-linear working range 0.5 nM to 5 μM
-LOD 0.5 nM;
-analysis of cultured human epidermal keratinocytes; |
[120] |
| 23. |
Superoxide dismutase; |
Cyclic voltammetry;
Electrochemiluminescence: Based on the signal (originating from the superoxide anion radical) emitted by methyl-cypridinalucifrin analogue at the electrode; |
-glassy carbon electrode modified with a composite consisting of ferrocene imidazolium salts and hydroxy-functionalized graphene, in a Nafion matrix; |
-supporting electrolyte: Phosphate buffer pH 5.3;
-peak accounting for superoxide dismutase activity present between 0.6 and 0.7 V in electrochemiluminescence, and between 0.7 and 0.8 in cyclic voltammetry;
-calibration plot linear in the 0.5 to 6.5 U·mL−1 SOD activity range, with LOD = 0.2 U·mL−1 in electrochemiluminescence; light emission lowered as SOD activity increased, correlated with oxygen generation from superoxide.
-RSD (n = 11) 2.3% for 2.0 U·mL−1 SOD in electrochemiluminiscence;
-scan range between 1.0 and −1.2 V.vs. Ag/AgCl in cyclic voltammetry and electrochemiluminescence
-scan rate: 0.1 V·s−1 |
[121] |
| 24. |
Glutathione; |
Cyclic voltammetry;
Amperometry;
Differential pulse voltammetry; |
-multiwalled carbon nanotubes@reduced graphene oxide nanoribbons core-shell heterostructure-modified glassy carbon electrode; |
-supporting electrolyte: 0.01 M phosphate buffered saline, pH 7.0;
-CV measurements carried out from−0.2 V to +0.6 V, or from +0.3 V to +0.8 V at a scan rate of 50 mV s−1, showing enhanced electrocatalytical activity for the developed electrode;
-DPV measurements carried out by scanning from +0.2 V to +0.7 V at a pulse amplitude of 50 mV;
-highest voltammetric response obtained at 0.55 V vs. Ag/AgCl;
-amperometric measurements carried out in 0.01 M phosphate buffer pH 7.0 at + 0.55 V vs. Ag/AgCl;
-LOD 0.039 μM (amperometry);
-two linear ranges: 0.05–266.3 μM and 266.3–766.3 μM (amperometry);
-RSD 3.53% (amperometry);
-analysis of real human serum samples; |
[122] |
| 25 |
Glutathione; |
Cyclic voltammetry;
Square wave voltammetry; |
-gold-copper metal-organic framework immobilized on the surface of a glassy carbon electrode; |
-supporting electrolyte: 0.1 mol L−1 phosphate buffer, pH 3.0;
-anodic oxidation peak appeared at around +0.30 V vs. Ag/AgCl in CV and +0.25 V vs. Ag/AgCl in SWV;
-linear dynamic range 1–10 μmol L−1 in SWV;
-LOD 0.30 μmol L−1 in SWV;
-sensitivity 0.89 ± 0.02 μA μmol L−1 in SWV;
-repeatability 2.14% in SWV;
-analysis of commercial tablets with more than 98% recovery; |
[123] |
| 26. |
Glutathione;
Glutathione disulphide; |
Cyclic voltammetry;
Square-wave voltammetry; |
-antimony trioxide–modified-carbon paste electrode; |
-supporting electrolyte: 0.2 mol/L Britton-Robinson buffer;
-oxidation potentials of +1.08 V for glutathione and +1.36 V for oxidized glutathione, vs. Ag/AgCl reference in CV and SWV;
-SWV responses linear in the concentration range of 2 to 300 μmol/L glutathione;
-LOD of 0.34 μmol/L glutathione and 0.1 μmol/L for oxidized glutathione in SWV;
-determination of glutathione and glutathione disulphide in urine samples;
-ascorbic acid, cysteine, glucose, glutamic acid and uric acid gave no significant interferences; |
[124] |
| 27. |
Uric acid; |
Cyclic voltammetry;
Chrono-amperometry; |
-glassy carbon electrode, modified with a ZnO/carboxylic acid/multiwalled nanotube composite; |
-supporting electrolyte: Phosphate buffer solution, pH = 7.0;
-cyclic voltammetric peak at 0.5 V vs. Ag/AgCl;
-chronoamperometric measurements performed at +0.577 V vs. Ag/AgCl;
-rapid current response time (<5 s);
-selective measurement of uric acid at clinically relevant concentrations (100–900 µM) by chronoamperometry; |
[125] |
| 28. |
Uric acid; |
Cyclic voltammetry;
Differential pulsevoltammetry; |
-ZnO nanorods and graphene nanosheets hybrid electrode sprayed on indium tin oxide (ITO) glass; |
-supporting electrolyte: Phosphate buffer saline, 0.01 M, pH = 7.4;
-potential range: −0.2 to 0.6 V, vs. Ag/AgCl reference electrode (CV and DPV);
-CVs were recorded at a scan rate of 50 mV s−1;
-cyclic voltammetric oxidation peak potentials of uric acid and ascorbic acid, were 0.36 V and 0.28 V, respectively,
-DPV analytical responses recorded with: A pulse height of 50 mV, a step height of 4 mV, a pulse width of 0.2 s, and a step time of 0.5 s;
-sensitivity for uric acid 0.3 μA μM−1 cm−2 (DPV);
-peak current intensities linearly related to the uric acid concentration in the range of 5–80 μM (DPV);
-LOD 5 μM (DPV);
-potentially applicable to clinical determination of uric acid; |
[126] |
| 29. |
Uric acid; |
Cyclic voltammetry; |
uricase/carboxymethylcellulose dispersed carbon nanotube/gold thin film biosensor; |
-supporting electrolyte: 0.05 M phosphate buffer solution (pH 7.4);
-cyclic voltammetric sweep rate: 50 mV s−1;
-sensitivity of 233 μA mM−1 cm−2 at +0.35 V vs. Ag/AgCl reference;
-linear range 0.02–2.7 mM;
-detection limit of 2.8 μM;
-detection of uric acid in serum and urine;
-negligible interferents effect from urea and ascorbic acid at physiological amounts; |
[127] |
| 30. |
Bilirubin; |
Cyclic voltammetry; Amperometry; |
-carbon electrode modified with multiwalled carbon nanotubes or electrochemically reduced graphene oxide; |
-supporting electrolyte: 0.1 M phosphate buffer solution (pH 7.2);
-scan rate of 50 mV s−1;
-two cyclic voltammetric oxidation peaks: At +0.25 V, corresponding to the oxidation of bilirubin to biliverdin and another at +0.48 V, corresponding to the oxidation of biliverdin to purpurine;
-graphene type electrode: Amperometric linear range 0.1–600 µM and LOD 0.1 ± 0.018 nM; sensitivity 30 nA µM−1 cm−2, at 0.48 V vs. Ag/AgCl reference;
-multiwalled carbon nanotube type: Amprometric linear range 0.5–500 μM; LOD 0.3 ± 0.022 μM; sensitivity 15 nA µM−1 cm−2, at 0.48 V vs Ag/AgCl reference;
-no interferences from glucose, ascorbic acid, uric acid, and glutathione;
-analysis of blood serum samples; |
[128] |
| 31 |
Bilirubin; |
Linear sweep voltammetry; Differential pulse voltammetry; |
-disposable screen-printed carbon electrodes obtained using graphite carbon ink printed on a PET substrate; |
-supporting electrolyte: 0.05 M Trizma buffer, pH 8.5;
-LSVs were recorded at a potential window of 0 to 0.6 V at a scan rate of 0.1 V/s;
-DPVs were obtained at a potential window of 0 to 0.6 V with pulse amplitude of 0.05 V and pulse width of 0.05 s;
-two anodic voltammetric peaks noticed on DPVs at around 0.25 V and 0.35 V (vs Ag/AgCl reference) corresponding to the oxidation of bilirubin to biliverdin, and of biliverdin to purpurin;
-linear range 5–600 µM (LSV);
-sensitivity 95 μA μM−1 cm−2 (LSV);
-good selectivity in the presence of glucose, creatinine and ethanol;
-application to serum samples; |
[129] |
| 32. |
Bilirubin; |
Cyclic voltammetry;
Differential pulsevoltammetry; |
-carbon paste electrode; |
-supporting electrolyte: 0.05 M phosphate buffer solution (pH 8.0);
-two step oxidation process at around 300 mV and around 500 mV (CV);
-DPV parameters: Pulse time 10 ms, potential step 5 mV and 150 ms optimized pulse amplitude;
-linear range 3.5–25 µmol L−1 in DPV, considering the signal of irreversible anodic oxidation at 320 mV vs. Ag/AgCl;
-LOD 1.2 µmol L−1 in DPV; |
[130] |
| 33. |
Melatonin; |
Cyclic voltammetry;
Fixed-potential amperometry; |
-screen-printed carbon electrode modified with graphene; |
-supporting electrolyte: 0.1 M phosphate buffer (pH 7.0);
-oxidation CV peaks at 0.22 V and 0.80 V; reduction peaks at 0.12 V and 0.75 V vs pseudosilver/silver chloride reference electrode,
-linear range of 1–300 μM in amperometry, at 0.8 V;
-LOD 0.87 × 10−6 M and LOQ 2.91 × 10−6 M in amperometry;
-RSD = 1.24% at the assay of Bien Dormir tablets (CV); |
[131] |
| 34. |
Melatonin; |
Square wave voltammetry; |
-glassy carbon electrode; |
-linear concentration range of 5–200 µM;
-LOD of 0.3432 µM;
-analytical peak present at about 650 mV;
-determination in pharmaceutical formulations and in human urine; |
[132] |
| 35. |
Melatonin; |
Square wave voltammetry; |
-carbon fiber microelectrode; |
-reliably quantified melatonin concentrations in the visual cortex of anesthetized mice after intraperitoneal injections of different melatonin doses;
-SWV enabled sensitive detection of oxidation peak at about 0.7 V vs. Ag/AgCl, discriminating melatonin from most common interferents; |
[133] |
| 36. |
Coenzyme Q10; |
Differential pulse voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Acetic acid containing 20% acetonitrile and 0.5 M CH3COONa;
-DPV pulse amplitude of 20 mV, scan rate of 20 mV s−1 and pulse width of 80 ms allowed both fast recording and good resolution;
-well-configured DPV cathodic peak attributed to reduction of CoQ10 at −20 mV vs. silver chloride external reference
-LOD 0.014 mM (12 mg L−1);
-LOQ 0.046 mM (40 mg L−1);
-linearity up to 1 mM, with excellent corelation (r = 0.9989);
-determination in commercial capsules; |
[134] |
| 37. |
conzyme Q10; |
Direct current voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Phosphate buffer solution (pH 6.86);
-reversible oxidation peak at +0.4 V, corresponding to oxidation of hydroquinone group; reduction peak at −0.6 V vs. silver chloride reference;
consistent to the previously confirmed reduction of ubiquinone to ubiquinol;
-linear range 2.0 × 10−5–2.0 × 10−4 M;
-assay of coenzyme Q10 in pharmaceuticals; |
[135] |
| 38. |
Coenzyme Q10;
α-lipoic acid; |
Cyclic voltammetry;
Square wave anodic stripping voltammetry; |
-MnO2-modified screen-printed graphene electrodes; |
-determinations performed in 20:80 (v/v) ratio of ethanol/acetate buffer 0.1 M at pH 4.0;
-the anodic peak of alpha lipoic acid present at a potential of 0.64 V, and that of coenzyme Q10 at 0.22 V, vs Ag/AgCl paste reference electrode (CV);
-optimized square wave parameters: 5 mV step potential, 20 mV amplitude, and 25 Hz frequency;
- linear range 2.0–75.0 μg mL−1 for coenzyme Q10, and 0.3–25 μg mL-1 for α-lipoic acid in square wave anodic stripping voltammetry;
-LOD 0.56 μg mL−1 for coenzyme Q10 and 0.088 μg mL−1 for α-lipoic acid in square wave anodic stripping voltammetry;
-determination in dietary supplements with good specificity in the presence of other vitamins and ionic species; |
[136] |
| 39. |
Vitamin D2 and D3; |
Cyclic voltammetry;
Differential pulsevoltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: 40% ethanol/60% water containing LiClO4;
-potential range of 0.0 to +1.5 V and scan rate of 50 mV s−1 (CV);
-three well-configured, separate DPV peaks: Vitamin D around 0.594 V, vitamin E around 0.334 V, and vitamin A around 0.841 V vs. Ag/AgCl reference;
-LOD 1.3 × 10−7 (vitamin D2) and 1.18 × 10−7 mol/L (vitamin D3) in DPV;
-determination in vitamin D3 tablets; |
[137] |
| 40. |
Vitamin D2 and D3; |
Cyclic voltammetry;
Differential pulse
voltammetry; |
-glassy carbon electrode modified with AuPd; |
-supporting electrolyte: Ethanol/water (40%/60%: v/v) containing lithium perchlorate;
-CV scan rate of 50 mV/s, in the domain 0.0–1.5 V;
-DPV scan rate: 10 mV/s, sampling time: 20 ms, pulse interval: 100 ms.
-detection potential of +0.4 V vs. Ag/AgCl enabled diminution of interferences and good separation from vitamins A and E (DPV);
-linear ranges 1–10 μM vitamin D2, 5–50 μM vitamin D3 in DPV;
-LOD 0.15 μM vitamin D2 and 0.18 μM vitamin D3 (DPV);
-detection of vitamin D3 in drug specimen; |
[138] |
| 41. |
Vitamin D3; |
Square wave
voltammetry; |
-boron-doped diamond electrode; |
-supporting electrolyte: 0.02 mol L−1 Britton-Robinson buffer pH 5.0 prepared in 50% ethanol;
-well-defined voltammetric peak at around +1.00 V vs. Ag/AgCl;
-linear range 2 to 200 mol L−1;
-LOD 0.17 μmol L−1;
-LOQ 0.51 μmol L−1;
-determination in pharmaceutical products; |
[139] |
| 42. |
Alpha lipoic acid; |
Cyclic voltammetry;
Chronoamperometry;
Differential pulse voltammetry; |
-pyrolytic graphite electrode modified with cobalt phthalocyanine; |
-supporting electrolyte: Phosphate buffer solution (pH = 7.0);
-scan rate (CV) 25 m Vs.−1;
-oxidation peak present at 0.84 V vs. SCE (CV), with highest response resulted from modification with cobalt phthalocyanine;
-LOD 2.5 × 10−7 mol L−1 and LOQ 8.3 × 10−7 mol L−1 (CV);
-LOD 9.8 × 10−8 mol L−1 and LOQ 3.2 × 10−7 mol L−1 (Chronoamperometry);
-LOD 3.4 × 10−9 mol L−1 and LOQ 1.2 ×1 0−8 mol L−1 (DPV);
-determination in pharmaceutical dietary supplement samples; |
[140] |
| 43. |
Alpha lipoic acid; |
Amperometry; |
-cobalt phthalocyanine–modified pyrolytic graphite electrode, integrated in a batch injection analysis set-up; |
-supporting electrolyte: 0.1 mol L−1 phosphate buffer, pH 7.0;
-applied potential of 0.9 V vs. Ag/AgCl;
-linear response in the range 1.0 × 10−5–1.3 × 10−4 mol L−1;
-LOD 1.5 × 10−8 mol L−1;
-quantification in dietary supplements and in synthetic urine; |
[141] |
| 44. |
Alpha lipoic acid; |
Cyclic voltammetry;
Differential pulse voltammetry; |
-SnO2 nanoparticles-modified glassy carbon electrode; |
-supporting electrolyte: Britton-Robinson buffer pH 4.5;
-well-defined DPV oxidation peak at 0.843 V vs. Ag/AgCl;
-two linear dynamic ranges of 0.50–50 and 50–400 μmol L−1 (DPV);
-LOD 0.13 μmol L−1 (DPV);
-LOQ 0.43 μmol L−1 (DPV);
-analysis of pharmaceutical dosage forms, with RSD between 0.45 and 6.2%; |
[142] |
| 45. |
Tert-butylhydroquinone and butylated hydroxyanisole |
Cyclic voltammetry; Square-wave voltammetry; |
-carbon black paste electrode; |
-optimum conditions of electrolyte: 0.2 mol L−1 phosphate buffer (pH 7.0), 600.0 μmol L−1 surfactant cetylpyridinium bromide;
-scan rate 50.0 mVs−1;
-anodic cyclic voltammetric peak at cca 0 V vs. Ag/AgCl for tert-butylhydroquinone and at 0.4 V for butylated hydroxyanisole;
-LOQ for tert-butylhydroquinone 0.23 μmol L−1 (SWV) and 0.27 μmol L−1 (DPV);
-LOQ for butylated hydroxyanisole 0.26 μmol L−1 (SWV) and 0.23 μmol L−1 (DPV);
-determination in mayonnaise, margarine, biodiesel; |
[143] |
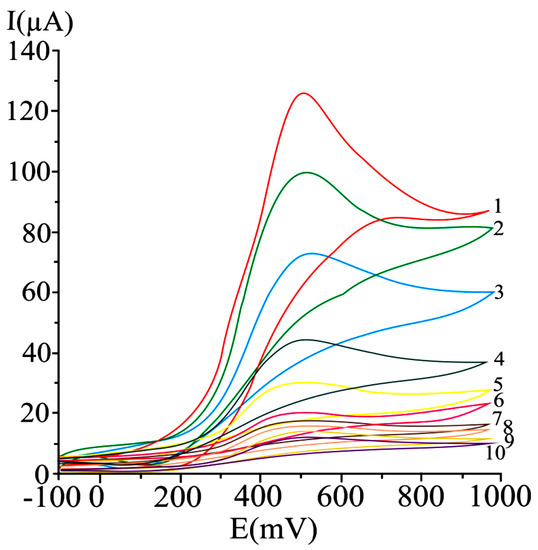
A series of irreversible cyclic voltammograms recorded for increasing ascorbic acid concentrations, at a carbon paste electrode are given in .
Figure 1. Cyclic voltammograms obtained at a carbon paste working electrode at varying ascorbic acid concentrations, as mM: 20 (1), 15 (2), 10 (3), 5 (4), 2.5 (5), 1.25 (6), 0.625 (7), 0.31 (8), 0.15 (9), and 0.07 (10); potential scan rate 50 mV/s [99].
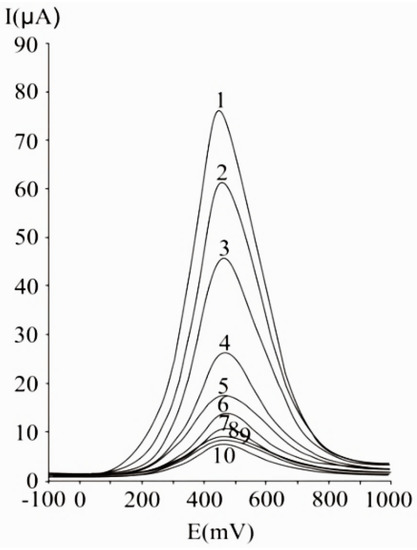
A 50 mV s−1 cyclic voltammetric scan rate could enable analyte diffusion to the electrode and promoted electron transfer. In the case of irreversible or quasi-reversible voltammograms [99][144], at elevated scan rates, electron transfer is slow relative to the applied potential sweep rate, so the rate of establishing the equilibrium at the electrode is diminished. In the case of very elevated scan rates, peaks have high current intensities, but distortions may occur on the voltammogram [144]. In differential pulse voltammetry at carbon paste electrode (), the optimum value of 75 mV was used for the pulse amplitude, and 125 ms was chosen for the pulse period, allowing a good compromise between promoting analytical signal, minimizing noise and good resolution [99].
Figure 2. Differential pulse voltammograms obtained with a carbon paste working electrode for different ascorbic acid concentrations, expressed as mM: 20 (1), 15 (2), 10 (3), 5 (4), 2.5 (5), 1.25 (6), 0.625 (7), 0.31 (8), 0.15 (9), and 0.07 (10); experimental conditions: Pulse amplitude 75 mV, pulse period 125 ms, potential scan rate 50 mV/s [99].
4. Determination of Total Antioxidant Activity with Carbon-Based Electrodes
Bare and modified carbon-based electrodes have been used for the antioxidant activity and its main contributors’ assessment. The electrochemical measurements are performed as per reference to a standard antioxidant, Trolox, gallic acid, ascorbic acid or quercetin, etc. In most of the cases, the peak intensities or peak areas are considered, providing quantitative informations.
Often, authors report indexes illustrating antioxidant activity: IC30 and IC50 [145]. IC50 corresponds to the antioxidant amount that induces a change of a model signal by 50%. The lower the concentration required for 50% increase or inhibition of the considered model signal, the more active the antioxidant. Korotkova et al. [146] relied on the modifications in the oxygen electro-reduction current in the presence of antioxidants, as criteria to determine IC50 by voltammetry. Catalase and superoxide dismutase yielded an increase of the oxygen electro-reduction current, when compared to the signal obtained in supported electrolyte. Superoxide dismutase presented an IC50 value of 1.08 μM [146]. Wei et al. evaluated IC50, as the antioxidant amount that diminishes the oxidation peak of superoxide by 50%. They reported an IC50 value of ascorbic acid of 5 × 10–4 mol/L [147].
Blasco et al. [148] define the “Electrochemical Index” as the total polyphenolic content measured by an electrochemical technique. In this approach, the corresponding total phenolics concentration obtained from the “total electrochemical signal” was named “Electrochemical Index”. The total electrochemical signal was assimilated to the total amperometric current measured at controlled potential (800 mV), to achieve oxidation of all polyphenolics at neutral pH of 7.5. So, the “Electrochemical Index” became an approach to “total polyphenols” in samples without ascorbic acid (or alpha-tocopherol), or an approach to “total natural antioxidant” content in those samples where ascorbic acid (or alpha-tocopherol) could be present [148].
An overview of the analytical parameters and real sample applications regarding the assay of total antioxidant activity by the use of carbon-based working electrodes is given in [149][150][151][152][153][154][155][156][157][158][159][160][161][162][163][164][165][166][167][168][169][170][171][172][173][174][175][176][177][178][179][180][181].
Table 3. Some relevant examples of electrochemical assay of antioxidant activity and its key contributors [149][150][151][152][153][154][155][156][157][158][159][160][161][162][163][164][165][166][167][168][169][170][171][172][173][174][175][176][177][178][179][180][181].
| No. |
Analytical Method |
Carbon-Based Working Electrode |
Analytical Characteristics |
Ref. |
| 1. |
Cyclic voltammetry;
Chronoamperometry;
Square-wave
voltammetry; |
-poly(gallic acid)/multiwalled carbon nanotube modified glassy carbon electrode; |
-supporting electrolyte 0.2 M H3PO4;
-cyclic voltammetric scan rate 50 mV/s;
-catalytic rate constant of 2.75 × 104 mol L−1 s−1, in chronoamperometry;
-voltammetric oxidation peak for gallic acid at 0.53 V vs Ag/AgCl, in CV and SWV;
-linear range of 4.975 × 10−6 to 3.381 × 10−5 M (SWV);
-LOD 3.22 × 10−6 M gallic acid (SWV);
-the SWVs of a fresh pomegranate juice sample shows three anodic peaks at 0.60, 0.70 and 1.0 V; signals can be attributed to the oxidation of different polyphenolic compounds, including gallic acid and catechin;
-determination of total phenolic content in pomegranate juice, as gallic acid equivalent;
-lack of interference of ascorbic acid, fructose, potassium nitrate and barbituric acid; |
[149] |
| 2. |
Cyclic voltammetry;
Differential pulse voltammetry; |
-nanocarbon-nanosilver hybrid electrode; |
-supporting electrolyte: Phosphate buffer solution, pH 7.0;
-CV studies confirmed that silver nanoparticles were efficiently immobilized on the Printex carbon surface; anodic and cathodic peak potentials noticed, were assigned to the redox pair Ag0/Ag+, whose presence was confirmed in the nanocomposite’s structure;
-DPV peak of gallic acid at 0.091 V vs. Ag/AgCl;
-sensitivity 0.254 μA/mol L−1 in DPV;
-LOD 0.0663 μM in DPV;
-linear range 5.0 × 10−7–8.5 × 10−6 in DPV;
-estimation of antioxidant activity in wine; |
[150] |
| 3. |
Cyclic voltammetry;
Amperometry; |
-single-walled carbon nanotubes electrode, covalently functionalized with polytyrosine; |
-supporting electrolyte 0.050 M phosphate buffer solution, pH 7.40;
-CVs recorded between −0.200 V and 0.800 V (vs. Ag/AgCl) at a scan rate of 0.100 V s−1;
-cyclic voltammetric oxidation peak potential for gallic acid at 0.2 V;
-amperometric working potential 0.200 V;
-amperometric sensitivity 163.2 mA/mol L−1;
-amperometric LOD 8.8 × 10−9 M;
-quantification of polyphenols in tea extracts: Green-Patagonia, red-Patagonia, classic-Green Hill and herbal (Taragüí); |
[151] |
| 4. |
Differential pulse
Voltammetry; |
-TiO2 nanoparticles/ multiwalled carbon nanotubes-modified glassy carbon electrode;
-guanine biosensor based on TiO2 nanoparticles and multiwalled carbon nanotubes, immobilized on glassy carbon electrode; |
-supporting electrolyte:
phosphate buffer solution, pH 7.4;
-oxidation DPV peak at 0.80 V (vs. SCE) corresponding to the electro-oxidation of guanine at the developed biosensor;
-the peak intensity value of guanine oxidation increased linearly with increasing metabisulfite (employed as OH radical scavenger) concentration from 1 to 30 mmol L−1;
-LOD 0.54 mmol L−1 for the guanine biosensor;
-quantification of the antioxidant capacity in drug samples (adrenaline hydrochloride injection); |
[152] |
| 5. |
Cyclic voltammetry;
Differential pulse
Voltammetry; |
-carbon paste electrode; |
-supporting electrolyte: 0.1 M phosphate buffer, pH 5.0;
-a cyclic voltammetric anodic peak at 0.33 V, with a corresponding cathodic peak at 0.28 V, vs Ag/AgCl, for 1% coffee sample in 0.1 M phosphate buffer pH 5.0;
-two further anodic peaks at 0.55 V and 0.78 V were observed in DPVs of 0.5% coffee sample, in the same electrolyte;
-good correlation with DPPH photometry and HPLC;
-evaluation of the antioxidant activity of roasted coffee samples;
-determination of electrochemical index of roasted coffee samples on the basis of the sum of the ratios of anodic peak currents to anodic peak potentials noticed on DPVs; |
[153] |
| 6. |
Cyclic voltammetry;
Square-wave voltammetry;
Chronoamperometry |
-nanocomposite-graphene/poly (3,4-ethylenedioxythiophene): Poly (styrenesulfonate) modified screen-printed carbon electrode; |
-supporting electrolyte: Ethanolic phosphate buffer solution based on 60% ethanol and 0.1 M phosphate buffer saline, pH 7.0;
-method relied on DPPH reduction by antioxidants;
-the presence of Trolox yielded a well–countoured anodic peak at around 0.9 V and a small cathodic peak at 0.3 V.
-cathodic cyclic voltammetric peak potentials of catechin and caffeic acid were present at −0.03 V and−0.025 V vs. Ag/AgCl;
-square voltammetric peak of DPPH at 0.25 V;
-chronoamperometric DPPH detection at 0.2 V vs. Ag/AgCl; the linear calibration between the difference of cathodic DPPH currents (in the presence and absence of standard Trolox solution) and Trolox concentration in a range of 5–30 μM;
-LOD 0.59 μM and LOQ 1.97 μM (chronoamperometry);
-RSD of reproducibility is 2.13% (chronoamperometry);
-RSD of repeatability 2.78% (chronoamperometry);
-evaluation of the antioxidant activity in Thai herb and herbal beverage, expressed as mg of Trolox/g of sample; |
[154] |
| 7. |
Differential pulse voltammetry; |
-multi-walled carbon nanotubes-modified glassy carbon electrode; |
-supporting electrolyte: 0.1 M phosphate buffer (pH 4.0–7.0);
-three DPV oxidation peaks observed at 0.39, 0.61 and 0.83 V for red dry wine and at 0.39, 0.80 and 1.18 V vs. Ag/AgCl for white dry wine, in phosphate buffer pH 4.0;
-RSD% (as gallic acid equivalents) comprised between 1.0 and 6.9, as function of the wine sample;
-evaluation of red and white dry wine antioxidant capacity, as gallic acid equivalents per 1 L of wine; |
[155] |
| 8. |
Differential pulse voltammetry; |
-carbon paste electrode;
-laccase-based modified carbon paste biosensor for the determination of phenolic content; |
-supporting electrolyte: 0.1 mol L−1 phosphate buffer, pH 6.0;
-biosensor was characterized by enhanced activity in mild acid medium and the response time (corresponding to the time required for enzyme oxidation of phenolic compounds), was lower than 30 s, but gradually increased up to 240 s, when a plateau was reached;
-honey samples presented 2 to 3 anodic DPV peaks, the first at about 0.2 V, the second at about 0.5 V and the third nearby 0.8 V vs. Ag/AgCl;
-electrochemical index determination, based on the sum of ratios of peak currents to peak potentials;
-determination of phenolic content in honey samples; |
[156] |
| 9. |
Cyclic voltammetry;
Differential pulse voltammetry; |
-carbon black nanoparticles press imprinted films; |
-supporting electrolyte: Phosphate buffer pH 7.40;
-scan rate of 50 mV s−1 in the potential range of -0.20 V to +1.0 V vs. Ag/AgCl (CV);
-pulse amplitude 50 mV/s, scan rate 10 m Vs−1 (DPV);
-anodic peaks of o-diphenols and m-phenols present in olive oil extract, noticed in the range 0.120–0.160 V and 0.590–0.610 V (vs. Ag/AgCl), respectively; consistency with results obtained for the standards (DPV);
-good repeatability for o-phenols;
-RSD < 6% (o-phenols), RSD < 15% (m-phenols) in CV;
-stable and reproducible voltammetric response of carbon black nanoparticles-based electrode;
-determination of phenolic content and electrochemical indexes in olive oil extracts, using hydroxytyrosol and tyrosol as standards; |
[157] |
| 10. |
Cyclic voltammetry;
Differential pulse voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Dimethylsulfoxide, with tetrabutylammonium hexafluorophosphate 0.1 mol L−1;
-the CVs were obtained at a scan rate of 100 mV s−1;
-DPV pulse width = 5 mV, pulse amplitude = 60 mV and scan rate = 20 V s−1;
-oxidation of ascorbic acid at 0.90 V in cyclic voltammetry and around 0.75 V vs. Ag/AgCl in differential pulse voltammetry;
-in the CVs of the bark extract, a very well contoured peak was observed at 1.3 V, corresponding to meta-diphenols and isolated phenols;
-in the CVs of the root and leaf extracts, an additional peak at 0.9 V indicates the presence of phenolics with ortho- or para-diphenol groups, in low amounts;
-determination of the antioxidant capacity of Bunchosia glandulifera (Jacq.) Kunth (Malpighiaceae) extracts, using ascorbic acid as standard; |
[158] |
| 11. |
Chrono-amperometry;
Differential pulse voltammetry; |
-glassy carbon electrode modified with multi-walled carbon nanotubes;
-glassy carbon polyquercetin-modified electrode; |
-supporting electrolyte: Phosphate buffer pH 7.0;
-antioxidant capacity using gallic acid as reference;
-DPVs recorded from 0 to 0.8 V (pulse amplitude 50 mV, pulse time 50 ms and potential scan rate 10 mV/s);
-DPVs of tea on the polyquercetin-modified electrode exhibited oxidation peaks at 0.080 and 0.19 V depending on the type of tea and a less configured oxidation step between 0.55 and 0.62 V vs Ag/AgCl;
-chronoamperograms recorded at a constant potential of 0.2 V, potential corresponding to oxidation of tea antioxidants;
-RSD = 0.5–20%, as function of the tea type (chronoamperometry);
-determination of the antioxidant capacity of tea, highest content for Green Sencha; |
[159] |
| 12. |
Cyclic voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Phosphate buffer pH = 7.0;
-cyclic voltammetric scans performed between 0.0 and 0.5 V vs Ag/AgCl at a scanning rate of 5 mV/s;
-anodic peak at 244 mV for pomace and its parts (skins and stems), and 252 mV for seeds;
-analyse of winemaking by-products (pomace, skins, seeds and stems separated from pomace); |
[160] |
| 13. |
Cyclic voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: 0.1 M sodium acetate–acetic acid buffer at pH 3.6;
-all the grapes revealed peak I at 0.26–0.31 V, peak II between 0.42 and 0.55 V, and peak III at approximately 0.66 V vs Ag/AgCl;
-correlations of anodic peak area with phenolic content and antioxidant activity were assessed;
-determination of phenolic contents and antioxidant capacity in 12 grape cultivars; |
[161] |
| 14. |
Cyclic voltammetry;
Square-wave
voltammetry;
Differential pulse voltammetry |
-glassy carbon electrode;
-laccase-modified carbon paste electrode; |
-supporting electrolyte: 0.1 M phosphate buffer solution, pH 6.0, using Ag/AgCl reference;
-CV: Scan rate of 100 mV s−1 within the range 0–1.4 V;
-SWV: Pulse amplitude 50 mV, frequency 50 Hz and a potential increment of 2 mV, scan rate of 100 mV s−1;
-first peak present between 100 and 400 mV, second between 0.55 and 0.7, and third at around 1 V, in CV/SWV;
-solutions of the extracts yielded highest DPV peaks at 0.2 V, alongside peaks present at 0.6 and 0.9 V;
-electrochemical indexes were calculated based on the sum of ratios of peak currents to peak potentials in DPV;
-antioxidant activity evaluation of dried herbal extracts;
-highest electrochemical indexes obtained for Gingko biloba and Hypericum perforatum, consistent with the results obtained by spectrophotometry; |
[162] |
| 15. |
Cyclic voltammetry;
Differential pulse voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte 0.1 M KCl;
-CV scan from 0 to +1000 mV at a scan rate of 100 mV s−1;
-DPV scan from 0 to +1000 mV at a scan rate of 100 mV s−1;
-the first peak of mature-phase milk occurred at around 400 mV; colostrum, had oxidation peaks at very high potential, around 800 mV (DPV) vs Ag/AgCl;
-mature-phase milk yielded a peak at around 400 mV; pasteurized milk had a peak at around 500 mV (CV);
-areas below oxidation peaks proportional to the amount of antioxidant compounds;
-free radical scavenging activity was highest for fresh breast milk and lowest for pasteurized breast milk, confirming the results obtained in DPPH assay;
-correlation between DPV and CV (r = 0.602, p < 0.001); correlation between DPV and DPPH method (r = 0.339, p = 0.003); correlation between CV and DPPH method (r = 0.468 p < 0.000); |
[163] |
| 16. |
Cyclic voltammetry;
Differential pulse voltammetry; |
-screen-printed carbon electrodes; |
-supporting electrolyte 0.1 M HCl;
-in CV, the potential recorded between 0.0 V and +1.2 V, at 100 mV s−1 scan rate, using silver pseudo-reference electrode;
-optimum DPV parameters: 100 mV modulation amplitude, 10 mV step potential, 0.05 s modulation time and 0.5 s interval time;
-ortho-diphenols (oleuropein, hydroxytyrosol and caffeic acid) show one anodic peak between 0.5 and 0.6 V, and one cathodic peak between 0.4 and 0.6 V (CV); same compounds present an anodic peak at +0.5 V, in standard and real sample (DPV);
-ferulic acid gave an oxidation peak at higher potential (0.7 V), in the standard solution, whereas this signal was almost negligible in the real sample (DPV);
-tyrosol is oxidized at +0.93 V, alongside other mono-phenols (such as vanillic acid) that suffer oxidation around this potential value, in standard and real sample (DPV);
-LOD of 0.022 mg L−1 for caffeic acid and tyrosol, in DPV;
-determination of hydrophilic phenols in olive oil; |
[164] |
| 17. |
Cyclic voltammetry;
Differential pulse voltammetry;
Square-wave voltammetry; |
-electroactivated pencil graphite electrode; |
-supporting electrolyte: 0.05 mol L−1 potassium hydrogen phthalate;
-naringenin is irreversibly oxidized, giving rise to two pH-dependent peaks due to mixed (diffusion- and adsorption-controlled) electrode processes involving two electrons and one proton;
-LOD = 3.06 × 10−8 mol L−1, and LOQ = 1.02 × 10−7 mol L−1 for DPV, expressed as naringenin;
-LOD = 4.40 × 10−8 mol L−1, and LOQ = 1.11 × 10−7 mol L−1 for SWV, expressed as naringenin;
-application to determination of polyphenol content in citrus juice; |
[165] |
| 18. |
Amperometry;
Cyclic voltammetry; |
-disposable polyester screen-printed graphitic macroelectrodes; |
-supporting electrolyte: 1:1 (v/v) methanol: Ethanol mixture containing 0.05 mol/L−1 LiCl;
-CV scans between −0.3 and +1.0 V, scan rate 50 mV s−1, for DPPH 1 mmol/L−1 (in 50 mmol L−1 LiCl prepared in methanol:ethanol) and for DPPH in the presence of antioxidants;
-chlorogenic acid, caffeic acid, catechin and quercetin were oxidized between +0.7 and +0.9 V (CV);
-oxidation processes of tocopherol and BHT occurred at more positive potentials, around +1.0 V (CV);
-amperometric detection of DPPH remaining after reaction with antioxidants, at +0.1 V (vs. pseudo AgCl).
-analysis of edible oils; |
[166] |
| 19. |
Cyclic voltammetry;
Differential pulse voltammetry;
Linear sweep
voltammetry; |
-multi-walled carbon nanotube paste electrode; |
-supporting electrolyte: 0.02 M acetate-acetic acid buffer/4% methanol (pH 4.5);
-CV scan between 0 and 1.5 V, at 100 mV s−1;
-DPV pulse amplitude 50 mV and scan rate 100 mV s−1
-CV oxidation potential at 1.12 V;
-DPV oxidation potential at 1.19 V;
-the average Tafel slopes of mushroom extract was found to be 1.258 mV per decade, in LSV;
-assay of Morchella esculenta L. as ethnomedicinal food;
-obtained net electrochemical antioxidant power as 2.7 ± 0.12 mg per gram, using ascorbic acid as reference; |
[167] |
| 20. |
Cyclic voltammetry; |
-carbon paste electrode incorporating 2,2-diphenyl-1-picrylhydrazyl; |
-supporting electrolyte: Phosphate buffer solution 0.1 M, pH 7.0;
-potential ranges investigated: 0.00 V to −1.00 V; 0.60 V to −0.20 V and 0.45 V to 1.10 V, vs. Ag/AgCl;
-a peak potential of −833 mV, due to the irreversible reduction of the nitro functions on the phenyl group, present in the structure of DPPH;
-for tea extract analyzed, signals recorded in the potential window 0.4 V–1.1 V;
-tea extracts presented an anodic peak at about 0.8 V and a cathodic one at around 0.75 V; |
[168] |
| 21. |
Cyclic voltammetry;
Amperometry; |
-single walled carbon nanotubes-, graphene- and gold nanoparticles-based screen-printed electrodes; |
-supporting electrolyte: Sodium phosphate buffer solution 0.1 M, pH 7.0;
-assessment of the quenching capacity of plant extracts (Hippophae fructus and Lavandula Flowers) in the presence of H2O2 (chosen as model reactive oxygenated species);
-cyclic voltammograms reveal anodic peaks below 0.45 V vs Ag/AgCl, in the presence of extract;
-a marked cyclic voltammetric anodic peak at 0.09 V, and a small cathodic peak at 0.35 V noticed for lavender extracts;
-amperometric assay based on sensor’s sensitivity to H2O2 in the absence / presence of the extract;
-best sensitivity obtained at the gold nanoparticles-modified sensor: 6.43 ± 0.2 µA cm−2 mM−1;
-amperometric determinations at constant potential of 0.55 V, with linearity of 2 to 30 mM hydrogen peroxide:
-antioxidant capacity determination of hydrosoluble plant extracts; |
[17] |
| 22. |
Cyclic voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Tetrabutylammonium perchlorate, in dimethyl sulfoxide 99%;
-scan rate 25 mV/s in CV;
-the voltammograms of the figs and almond extracts presented redox signals at positive potentials, the anodic oxidation being noticed at 1.175 V and 1.218 V, respectively, vs saturated calomel reference;
-determination of antioxidant activity of dry fruits (almond, apricot, cashew, figs, peanut, pistachio, raisins, and walnut); |
[169] |
| 23. |
Cyclic voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Sodium acetate-acetic acid buffer (0.1 mol L−1, pH = 4.5) in acidified 80% methanol;
-analytical cyclic voltammetric signals for all target phenolic compounds present between 0 mV and 800 mV, at a scan rate of 100 mV s−1;
-cyclic voltamogramms of berry fruits presented anodic peaks between 310 mV (quercetin) and 0.756 mV (coumaric acid) vs. Ag/AgCl
-antioxidant capacity quantification relied on the area underneath the anodic peak, corresponding to the charge up to a potential value of 500 mV (Q500);
-evaluation of antioxidant activity of 15 berry samples (strawberries, blackberries, blueberries and red raspberries); |
[170] |
| 24. |
Amperometry; |
-glassy carbon electrode integrated in a flow injection system with sequential diode array and amperometric detection; |
-supporting electrolyte: Ethanol 12% v/v and tartaric acid 2 g/L, pH 3.6;
-amperometric determinations at 800 mV vs. Ag/AgCl;
-calibration curve over the range 0–0.19 mM gallic acid equivalents;
-determination of total polyphenol content and antioxidant activity of white, red wines and oenological tannins;
-total wine phenolic content between 1.08 and 15.4 mM gallic acid;
-concentration range 0.07–0.34 mM gallic acid obtained for tannin solutions; |
[171] |
| 25. |
Cyclic voltammetry;
Differential pulse voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Sodium acetate 0.1 mol L−1;
-two CV oxidation waves at potentials of 0.45 V and 0.84 V vs Ag/AgCl, pointing towards the presence in the extract of minimum two kinds of reducing species, or a single reducing species that can be oxidized by two stable intermediates;
-extracts showed no voltammetric waves in the range of reduction potentials, suggesting that the reducing species in the extract of Mimosa albida leaves can exhibit antioxidant potential;
-two oxidation waves noticed on DPVs, indicating the existence of two antioxidant compounds: One species with greater antioxidant capacity with oxidation potential at 0.34 V, and the other one with lower antioxidant power, at 0.79 V;
-oxidation signal for Mimosa albida-modified silver nanoparticles at +0.3792 V (CV);
-analysis of aqueous leaf extract of Mimosa albida and assay of antioxidant capacity of Mimosa albida-modified silver nanoparticles; |
[172] |
| 26. |
Differential pulse voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: Sodium phosphate buffer solution 0.1 M, pH 7.4;
-scan rate 50 mV/s; pulse period 35 ms; potential step 10 mV;
-at increasing amounts of added extract, DPV oxidation peaks were noticed, at approximately 0.270 V, 0.430V, and 0.880 V vs. Ag/AgCl;
-determination of the antioxidant capacity of the Greigia Sphacelata fruit; |
[173] |
| 27. |
Cyclic voltammetry;
Differential pulse voltammetry; |
-glassy carbon electrodes modified with carbon nanotubes and chitosan; |
-supporting electrolyte: Britton-Robinson electrolyte buffer, at pH 3.0;
-chicoric acid anodic peak at 0.610 ± 0.060 V and cathodic peak at 435 ± 0.055 V vs Ag/AgCl, at 300 mV s−1 scan rate;
-the intensities of oxidation and reduction currents linearly vary with the square root of the scanning speed, in cyclic voltammetry;
-DPVs showed oxidation peaks for caftaric acid at 0.505 ± 0.002 V, and for chicoric acid at 0.515 ± 0.001 V vs Ag/AgCl, which are consistent with the results obtained at the assay of pharmaceutical forms;
-determination of total polyphenol content and antioxidant activity of Echinacea purpurea extracts in 3 different pharmaceutical forms (capsules, tablets and tincture); |
[174] |
| 28. |
Staircase voltammetry; |
glassy carbon electrode; |
-supporting electrolyte-100 mM KNO3;
-staircase voltammograms recorded successively for 4 cycles between +1.0 and −1.2 V vs Ag/AgCl;
-scan rate 50 mV/s, starting and ending in +1.0 V;
-the half-wave potential (E1/2), or the potential corresponding to half the anodic peak current (Ipa) was considered; lower E1/2 values are correlated to higher antioxidant potential;
-the peak intensity or, more accurately the surface area under the oxidation peak, provided quantitative informations: Antioxidant concentration/antioxidant capacity;
-analytical peaks present between −0.6 and 0 V, and around 0.5 V;
-evaluation of antioxidant activity for teas, wines and (superfood) juices;
-antioxidant index calculated relying on the maximum charge of oxidation (Qmax), the standard potential of the oxygen evolution reaction (vs. Ag/AgCl) and the standard potential of hydrogen evolution reaction (vs. Ag/AgCl); |
[175] |
| 29. |
Cyclic voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte −0.1 M H2SO4 solution;
-scan rate investigated in the range of 20–160 mV s−1;
-dependence of charge under the anodic peak, on the concentration of tested red corn pigments, quantified in the region −0.2–1.2 V;
-CV for dark red corn seeds extract (1 mg mL−1) presents two anodic peaks at about 0.4 V and 0.65 V; a cathodic peak at the reverse scan, at about 0.2 V vs saturated calomel reference;
-evaluation of total phenolic and flavonoid contents in red corn; |
[176] |
| 30. |
Voltammetry; |
-carbon fiber ultramicroelectrodes; |
-linear relationship between anodic peak current and caffeic acid (reference antioxidant) concentration from 3.0 to 500 μmol L−1;
-repeatability illustrated by a RSD of 2.7%;
-sensitivity 12 μA L mol−1;
-Ag/AgCl electrode used as reference;
-LOD 0.41 μmol L−1;
-LOQ 1.26 μmol L−1;
-estimation of antioxidant capacity in three different wines, and in green and red grape samples; |
[177] |
| 31 |
Cyclic voltammetry;
Square-wave voltammetry; |
-carbon electrode modified with guanine-, polythionine-, and nitrogen-doped graphene; |
-determinations performed in PBS pH = 1.5;
-1.0 mg/mL−1 guanine solution as optimum for modification of the electrode;
-a pair of redox peaks found between 0 and 0.3 V in CV; peak currents increased with increasing scan times; a thin blue membrane formed on the surface of electrode, showed that thionine was successfully polymerized;
-oxidation peak for ascorbic acid at about 1.1 V (SWV);
-linear range for ascorbic acid (standard antioxidant) analytical response ranged from 0.5 to 3.0 mg L−1 in SWV;
-LOD 0.21 mg L−1 (SWV);
-RSD 3.1% (SWV);
-determination of antioxidant capacity of fruit juices (grape juice, guava juice, and orange juice) and jute leaves extract, ramie leaves extract, and hemp leaves extract; |
[178] |
| 32 |
Differential pulse stripping voltammetry (DPSV);
Cyclic voltammetry; |
-glassy carbon electrode modified with polyglycine; |
-supporting electrolyte: Britton-Robinson electrolyte buffer, pH 3.0;
-well-configured oxidation peak of quercetin (model antioxidant) occurs at around +460 mV, a corresponding cathodic peak being visible at 420 mV vs Ag/AgCl;
-peaks shifted towards less positive potentials when the scan rates increased from 20 to 400 mV/s in CV;
-oxidation peak current assigned to phenolic compounds of yam, at 430 mV, consistent to the peak potential of quercetin, on differential pulse stripping voltammograms;
-DPSV-pulse amplitude of 50 mV, pulse width of 500 ms;
-LOD 0.39 µg L−1 (DPSV);
-LOQ 1.39 µg L−1 (DPSV);
-electrode modification resulted in 3.15-fold increase of sensitivity, when compared to the bare glassy carbon;
-total antioxidant capacity of 0.1 kg of yam, obtained as 96.15 +/− 0.85 µg/L of equivalents quercetin at 95% confidence level;
-relative standard deviation of 0.88%; |
[179] |
| 33 |
Cyclic voltammetry;
Chronoamperometry; |
-biosensor based on laccase immobilized onto a gold nanoparticles/graphene nanoplatelets-modified screen-printed carbon electrode; |
-supporting electrolyte: Sodium phosphate buffer solution 0.1M, pH 7.0;
-potential range from −0.6 V to 1.2 V with a scan rate of 0.05 V/s and a step potential of 2.0 mV (CV);
-anodic and cathodic CV peaks of hydroquinone at 0.2 V vs Ag/AgCl and 0 V, respectively, at a scan rate of 0.05 V s−1;
-excellent electrocatalytic activity towards oxidation of hydroquinone at a potential of −0.05 V in hydrodynamic amperometry;
-linear range 4–130 µM (chronoamperometry);
-LOD 1.5 µM chronoamperometry)
-LOQ 5 µM (chronoamperometry);
-determination of phenolic antioxidant capacity in wine and blueberry syrup; |
[180] |
| 34. |
Cyclic voltammetry; |
-glassy carbon electrode; |
-supporting electrolyte: 0.1 M sodium acetate/acetic acid bufer solution, pH 3.6;
-potential scans performed from −0.4 V to 1 V vs Ag/AgCl at a scan rate of 25 mV s−1;
-total antioxidant capacity expressed as ascorbic acid equivalents;
-crude medicinal plant extracts exhibited an oxidation peak around 750 mV on cyclic voltammograms;
-medicinal plant extracts have less than 36 times smaller total antioxidant capacity, when compared to ascorbic acid;
-it was concluded that cyclic voltammetry and FRAP are recommended for flavonoid quantitation; |
[181] |
5. Critical Perspectives and Comparative Conclusions
Analytical parameters of carbon-based sensors, analyte oxidation–reduction steps and potentials are influenced by analyte chemical structure (number and position of –OH groups or other substituents), electrode type, its development and interaction with the antioxidant molecule, as well as working conditions (matrix characteristics such pH or presence of interferences).
Studies performed on ascorbic acid electro-oxidation reveal irreversible voltammograms, consistent with the reported mechanism describing electrochemically reversible electron transfer, and a subsequent irreversible chemical process. During the first step, the oxidation of ascorbic acid involves liberation of two electrons and two protons, yielding dehydroascorbic acid. This electron transfer is followed by an irreversible solvation reaction at pH < 4.0. At pH values smaller than the first pKa value of L-ascorbic acid (around 4.5), two protons are exchanged during the process, whereas at higher pH values (4.5–8.0), a single proton is released, giving ascorbate anion as electroactive compound [182][183]. These described oxidation/solvation steps are consistent with the variation of the peak potential with pH, noticed up to pH 8.0, at a gold electrode: A pH increase from strong acidic to mild acidic and neutral values, results in anodic peak displacement to more positive values. Reported mechanistics aspects underlying ascorbic acid oxidation at pH > 8.0 imply ascorbate anion oxidation to a diketolactone, which by dehydration gives dehydroascorbic acid, that eventually suffers isomerisation to an ene-diol oxidizable at higher potentials [183].
Studies performed at a glassy carbon electrode modified by single-walled carbon nanotube/zinc oxide, proved that ascorbic acid anodic peak current increased with the increase of pH from 2 to 5, and reached a maximal value for pH comprised between pH 4 and 5. Then, the peak current progressively diminished as the pH increased from 6 to 10 and then increased again, for pH values found between 11 and 12. This confirms the involvement of deprotonation step in the oxidation of ascorbic acid. The oxidation peak shifted towards less positive potential values as the pH increased from 2 to 13. On the irreversible voltammograms obtained at the modified electrode, the oxidation potential shifted by approximately 240 mV towards a lower potential and the peak current doubled, when compared to the bare glassy carbon electrode, confirming electrocatalytical effect in a diffusion-controlled process (linear dependence of the current intensity on the square root of scan rate was observed) [184].
At a p-phenylenediamine film–holes modified glassy carbon electrodes, the cyclic voltammetric oxidation peak current of ascorbic acid increased, as the pH increased from pH 2 to 5. A further pH increase of the buffer resulted in a decrease of the analytical signal. This observation is consistent with the described analyte-electrode interaction: Within the pH range for which the modifier film is positively charged and the analyte is found in anionic form, the interaction of the modified glassy carbon electrode with ascorbic acid promotes the redox signal. With increasing pH value, the film may adopt a negative charge (caused by adsorption of free OH−), causing rejection of the anionic form of ascorbic acid. Therefore, the pH value of 5.0 was chosen as optimum, privileging interaction between the cationic film of modifier and the anionic form of analyte [185].
The analysis of anthocyanins by cyclic voltammetry at pH 7.0 revealed for kuromanine (cyanidin-3-O-glucoside) chloride and cyanin (cyanidin-3,5-O-diglucoside) chloride a first reversible peak at around 300 mV, due to the oxidation of catechol moiety present on the B-ring. The oxidized molecule can be subject to further oxidation at higher potential values yielding a second oxidation peak corresponding to the oxidation of the 5,7-dihydroxyl structure of the A-ring (resorcinol moiety). Given the different sensitivities of the techniques, the second peak obtained in cyclic voltammetry is smaller than that obtained in differential pulse voltammetry [105].
Studies on flavonoids reconfirmed first oxidation of the most redox active -OH groups present on ring B. Oxidations of the –OH groups present at C3 on ring C, and on the ring A (in resorcinol group) occur at more positive potentials. Studies on delphinidin anthocyanidin firstly reveal two peaks corresponding to oxidative processes involving -OH groups at 3′,4′,5′ positions (ring B), followed by a third peak corresponding to oxidation of -OH present on position 3 on ring C, and a fourth corresponding to oxidation of -OH groups present at positions 5 and 7 on ring A (resorcinol moiety) [104].
Oxidation of most ortho-diphenols occurs at close potentials, while mono-phenols are oxidized at higher potentials. Phenolic compounds that possess two −OH groups in ortho position on the aromatic ring, are reversibly oxidized to ortho-quinones: They give first peaks appearing on voltammograms, consistent with enhanced electron donating ability typically assigned to catechol-like polyphenols, endowed with highest antioxidant power. Most monophenols are subject to irreversible oxidation, due to only one –OH group. Ortho-diphenols present in olive oil, such as caffeic acid, hydroxytyrosol, and oleuropein are subject to reversible oxidation due to the presence of two hydroxyl groups in ortho position, exhibiting one anodic peak and one corresponding cathodic peak at the reverse scan. Tyrosol was irreversibly oxidized generating one anodic peak, due to the oxidation of only one −OH group present on the benzene ring, with the absence of the corresponding cathodic peak. Ferulic acid, a cinnamic acid derivative presenting only one −OH group, has one oxidation peak, and a much less configured reverse cathodic peak [164].
A cyclic voltammetric study performed on wine phenolics confirmed first oxidation of catechol moieties. Catechol-containing hydroxycinnamic acids, as main phenolics present in white wines exhibit a first peak at around 480 mV, followed by polyphenols with more positive potentials (900–1000 mV) such as coumaric acid and their derivatives. High molecular mass compounds resulting from oxidation of original white wine phenolics during storage can contribute to IInd peak. Catechin-type flavonoids, oligomeric and polymeric tannins present in red wines give a first peak around 440 mV; the second peak at around 680 mV was assigned to malvidin, with slight contribution from other phenolics such as trans-resveratrol; second oxidation of the catechin-type flavonoids resulted in a third anodic peak for red wines at 890 mV [113].
Yakovleva et al. performed an electroactivity-based general classification of the phenolic compounds under study (benzenediols, phenolic acids, flavonoids) into groups: Quercetin, dihydroquercetin, and phenolic acids with two −OH groups at ortho positions (2,3-dihydroxybenzoic, protocatechuic, and caffeic acid) are subject to reversible electro-oxidation at pH 6.0, below 400 mV; polyphenols with –OCH3 substituents present oxidation peaks in the range 400–600 mV; monophenols such as monohydroxyphenolic acids are oxidized at lower rate, having anodic peak potentials greater than 600 mV; the electron-donating groups −OH, −OCH3 or −CH3 present on the aromatic ring, were confirmed to render studied phenols more oxidizable. Monohydroxylated phenolics with methoxy substituents could be faster oxidized by electron donation than monohydroxyphenols lacking methoxy groups. It was stipulated that the mechanism underlying this trend is the stabilizing effect of the methoxy groups in the phenoxyl radical formed after electron loss by phenolic compounds [112].
In a study focused on the electrochemical behavior of anthocyanins and anthocyanidins, comparing myrtillin chloride (that has a pyrogallol group on the B-ring) with oenin chloride (that has a hydroxyl group on C4′, placed in ortho position with respect to two methoxy groups), the methoxy groups could not impart the previously discussed oxidation facility, the reverse effect being noticed: Hydroxyl group from pyrogallol moiety on the B-ring (the case of myrtillin chloride) was oxidized with greater facility than the hydroxyl group placed in the ortho position with respect to two methoxyl groups (the case of oenin chloride). Moreover, the hydroxyl group found in the ortho-position with respect two methoxyl groups (in oenin chloride), was more oxidizable than the catechol group placed in the ortho position with respect to a single methoxy group (in petunidin chloride) [105].
Electrochemical studies on phenolic antioxidants are performed in acid media or, most of them, close to physiological pH values, to hamper irreversible conversion of polyphenols at alkaline values. Cyclic voltammograms recorded for some phenolic acids and flavones at pH higher than 8.0 were characterized by irreversibility, and the anodic oxidation potential could not be precisely assessed [112]. It was asserted that phenolic acids can suffer dimerization and polymerization, synchronous with their oxidation by one-electron loss [186]. An increase of the pH value was corroborated with a linear diminution of the oxidation potentials in the case of anthocyanins. Moreover, an enhanced adsorption of the oxidation products (with fouling the electrode surface) was also noticed, correlated to a dramatic anthocyanins’ oxidation peaks decrease during second scan, at all pH values [105].
During a study performed on major phenolics present in coffee, an inverse linear dependence of oxidation peak potential vs pH was observed until reaching the pKa value, with a slope of approximately 59.2 mV, close to the Nerstian theoretical value, describing an electron:proton transfer processes. On the differential pulse voltammograms obtained for 0.5% w/v coffee sample, highest peak currents were noticed at middle-acid pH values (ranging between 5.0 and 6.0), while the slope was broken and peak currents diminished at alkaline pH of 8.0. The same observation was valid for voltammograms of hydroxycinnamic acid derivatives. It was concluded that the redox behavior of electroactive species from coffee samples is dominated by catechol-like main redox contributors, endowed with most enhanced antioxidant features [153].
A series of electroanalytical methods have proved their viability in antioxidant and antioxidant activity assay, largely employed being cyclic, differential pulse and amperometric techniques. In differential pulse and square wave techniques, the influence of capacitive currents is hampered, promoting sensitivity and detection of irreversible oxidation–reduction processes linked to the presence of minor electroactive contributors. Moreover, linear scan (cyclic voltammetric) techniques proved their viability in the comparative study of antioxidants’ electroactivity (relying on peak position), as well as in antioxidant capacity assay (based on the area under the anodic peak).
Carbonaceous electrode properties affecting electrochemical response are represented by surface properties, electronic structure, adsorption, electrocatalytical behavior and surface preparation [187]. The performances of carbon electrodes as working electrodes depend on the structure of the electrode, on the chemical bonds established between the carbon atoms, as these factors influence mechanical, conductive and chemical features. Considering allotropic carbon forms, diamond, whose structure is ensured by sp3 bonds, has poor conductivity, thus requiring boron dopping. Carbon allotropic forms whose structure is ensured by sp2 bonds are endowed with convenient electrical features, mainly conductivity.
Electrochemical determinations rely on surface phenomena and thus, electrode surface represents an important characteristic influencing analytical performances, reaction kinetics and interactions with the compounds to be analyzed. Electrode surfaces are commonly polished using alumina, mainly in the case of glassy carbon. A significant voltammetric current increase was reported in the case of gallic acid, chlorogenic acid, caffeic acid, catechin, rutin and quercetin, as consequence of surface modification with alumina, by mere abrasive polishing [74]. It was found that modification of the glassy carbon electrode surface with small amounts of alumina can result in enhanced apparent catalysis of electron transfer involving catechol at low pH value. It was reported that catechol suffers adsorbtion on alumina, not directly on the electrode, so the process involves the triple boundary present between alumina particles, analyzed solution and the electrode. Enhancement of catechol redox process takes place at low pH, as such values facilitate the proton-coupled electron transfer at the previously-mentioned three-phase boundary [70]. The antioxidant capacity and activity, as well as the electrochemical index are significantly influenced by the presence of alumina in the case of tea, wine and phytotherapics samples. The improvement in detectability and sensitivity recommends the use of alumina-modified glassy carbon electrode for this assay. The shift of the anodic peak potentials towards less-positive values was reported, thus indicating that the electron transfer is promoted by surface modified with alumina. Nevertheless, the reported diminution in the difference between peak potentials (affecting peak-to-peak discrimination), may constitute a shortcoming in the case of surface modification with alumina [74].
Glassy carbon electrodes are prone to other modification methods: Electrode coating with ionic polymers was applied due to their ability to enhance conductive properties. A glassy carbon electrode coated with a thin film of poly(trihexylvinylbenzylammonium chloride) proved permselectivity to uric acid, that presented a linear voltammetric analytical response in the concentration range of 1–10 μM [188]. A glassy carbon electrode modified with multi-walled carbon nanotubes dispersed in polyhistidine allowed for simultaneous differential pulse voltammetric determination of ascorbic acid and paracetamol with improved analytical responses at minimized overvoltage and under diffusion control, benefiting from large electroactive area and enhanced electrocatalytical properties [189].
Electrolytes influence the surface of carbonaceous electrodes. Studies performed in sulfuric acid 4.5 M confirmed a change in composition of surface compounds, removal of finely crystalline graphite, as well as of unstable functional groups from the surface of activated carbon [190].
Conventional carbon-based sensors include glassy carbon, carbon fiber or pyrolytic graphite electrodes. Developing sensors at sizes below 100 nm is often a key characteristic that typically results in high surface area and improved surface/volume ratio. Other distinctive, attractive properties are swift electron transfer, enhanced electrocatalytic activity, improved interfacial adsorption and biocompatibility, when compared to other conventional materials [191][192]. Carbon-based nanomaterials composed of graphitic-type atoms linked by sp2 bonds, include fullerenes, carbon nanotubes and graphene. Their behavior depends on the interactions established with other materials and atomic or molecular structures [191][193][194][195][196]. These nanomaterials possess hollow or layered structures, and can establish non-covalent linkages with organic species through π-π stacking, hydrophobic interactions, hydrogen bonding, van der Waals or electrostatic forces [191][197].
Carbon nanomaterials are prone to combination with a series of other nanomaterials (based on noble metal nanocrystals, ceramics or Teflon) giving rise to nanocomposites with improved characteristics in a distinctive, novel material [191]. Previously reported novel application reveals that oxidation peaks of key analytes such as ascorbic acid, dopamine and uric acid at a ZnO nanosheet arrays/graphene foam sensor, are higher than those obtained at metal oxide-based (indium tin oxide) electrode, proving enhanced electrochemical features in the assay of both analytes, linked to the high conductivity of 3D porous graphene foam and large specific surface imparted by ZnO nanosheet arrays [198]. Diminished ascorbic acid overpotential and good peak-to-peak DPV separation for ascorbic acid vs dopamine (218.0 mV), dopamine vs paracetamol (218.0 mV), and ascorbic acid vs paracetamol (436.0 mV) were reported for platinum nanoparticles-decorated graphene nanocomposite electrode, with performances improved versus graphene-modified glassy carbon electrode and bare glassy carbon electrode [101]. A ZnO nanorods/graphene nanosheets/indium tin oxide hybrid electrode allowed good discrimination between uric acid and ascorbic acid, peaks at ZnO nanorods/indium tin oxide being not configured [126].
Carbon paste electrodes are synthesized by combining carbon-based materials (carbon nanotubes, microspheres or nanofibers, acetylene black, graphite and diamond,) with a binder (silicon oil or mineral oil) and largely used in antioxidant assay. A highly rigorous control over pasting liquid amount is required, as large amounts, although able to lower background currents, may diminish electron transfer rates [199][200]. These materials are easy to synthesize, inexpensive, possess reduced ohmic resistance, have broad potential range. They are prone to facile modification by using redox complexes, metal oxides or ionic surfactants, to improve performances in voltammetric assays [64]. The use of a carbon paste electrode modified with copper oxide-decorated reduced graphene enabled simultaneous voltammeric assay of ascorbic acid, uric acid and cholesterol, with sensitivity and good separation in SWV [103].
Employing printing technologies for rapid assay of electroactive species benefits from adaptability, compactibility, reduced costs, facility of large scale production, onsite application and easy modifier incorporation. Characteristics such as the employed ink material, the type of substrate, the choice of a particular electrochemical technique, the extent of waste generation, the obtained analytical performances, as well as shortcomings of each application of printed electrode should be considered. Researches should be focused on developing printed electrodes characterized by best analytical performances, lacking hazardous effects to the environment, to successfully replace conventional electrochemical cells [201].
The applications of chemical and biochemical carbon-based sensors encompass swift evaluation of key antioxidants and antioxidant activity in foodstuffs and dietary supplements, as well as in various biological media, these aspects being tightly related to screening/maintaining health status, hence the permanent interest in improving analytical performances and adaptability.
Electrode modification with single-, multiwalled carbon nanotubes or noble metal nanoparticles promotes electrocatalytical features, facilitating electron transfer: Peaks appear at lower potential and have higher corresponding current intensity. Hybrid nanosensors made of carbonaceous materials and metal/metal oxides enable for antioxidant signal separation and enhanced sensitivity in complex media.