 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jordan Holl | + 1740 word(s) | 1740 | 2021-03-26 04:13:43 | | | |
| 2 | Bruce Ren | -21 word(s) | 1719 | 2021-04-07 04:22:06 | | |
Video Upload Options
With the global prevalence of type 2 diabetes mellitus steeply rising, instances of chronic, hard-healing, or non-healing diabetic wounds and ulcers are predicted to increase. The growing understanding of healing and regenerative mechanisms has elucidated critical regulators of this process, including key cellular and humoral components. Despite this, the management and successful treatment of diabetic wounds represents a significant therapeutic challenge.
1. Introduction
The global impact of diabetes, including type 2 diabetes mellitus (T2DM), is severe, costing over 760 billion dollars—constituting 10% of adults’ annual health expenditure. More importantly, diabetes is projected to affect over 700 million individuals by 2045 (7.8% of the global population) [1][2]. In 2019 alone, more than 4 million adults died from direct and associated complications of diabetes. This prevalence and burden clearly outline diabetes and its associated complications as pressing global concerns.
Diabetic patients develop wounds characterized by impaired healing, prolonged inflammation, and reduced epithelization kinetics. Notably, 15% of patients suffering from T2DM develop ulcers localized on the lower limbs, referred to as diabetic foot ulcers (DFUs). DFUs represent the most severe form of diabetic wounds which may lead to lower limb amputation or death [3]. In fact, DFUs precede 84% of all diabetes-related lower limb amputations. Therefore, there exists a substantial need to elucidate the pathological processes causing ulceration, and which affect wound healing in diabetics.
Wound healing is defined as a natural physiological process occurring as the reaction to structural damage of tissues, including skin. These mechanisms involve sophisticated complimentary interactions between different cell types, acting through networks of soluble mediators, including cytokines, chemokines, growth factors, and metabolites. Wound healing consists of four subsequent and overlapping phases: hemostasis, inflammation, proliferation (re-epithelization), and remodeling (scar maturation).
Interestingly, diabetic hyperglycemia contributes to a variety of systemic complications, causing an array of local pathologies manifesting within the wound microenvironment, including chronic inflammation, dysregulated angiogenesis, hypoxia-induced oxidative stress, neuropathy, advanced glycation end-products, and impaired neuropeptide signaling [4].
2. Wound Healing Starts with Homeostasis
Immediately after wounding, degranulation of mast cells induces capillary permeability, in addition to vasodilation, increasing bleeding and allowing the influx of immune cells. Furthermore, the coagulation system is activated, and a scab is formed of provisional components [5]. Simultaneously, activated keratinocytes, fibroblasts, and platelets release soluble mediators: (a) growth factors, such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and vascular endothelial growth factor (VEGF); (b) chemokines, including IL-8 (CXCL-8) and CXCL-2; (c) danger-associated molecular patterns (DAMPs) such as histones, genomic DNA, adenosine 5′-triphosphate (ATP), high mobility group box protein 1 (HMGB1); and (d) cytokines; namely, thymic stromal lymphopoietin (TSLP), IL-33, and IL-25 [6][7][8][9]. Notably, all of the above-mentioned inflammatory mediators act as danger signals. Consequently, they trigger the infiltration of patrolling inflammatory cells and the induction of local immune responses (inflammation phase) and subsequent proliferative induction of tissue-resident cells.
3. Wound Inflammation Orchestrates Healing and Regeneration
A strong inflammatory cascade, initiated during hemostasis, now commences to clean the wound of debris, damaged cells, and microbes. The inflammatory phase is characterized by (a) an influx of inflammatory cells including neutrophils, monocytes/macrophages, mast cells, and T cells; (b) the accumulation of inflammatory mediators such as cytokines, chemokines, and lipid mediators; and c) the release of extracellular matrix degradation enzymes such as matrix metalloproteases (MMPs) and collagenases; causing swelling, heat, and pain (Figure 1) [4].
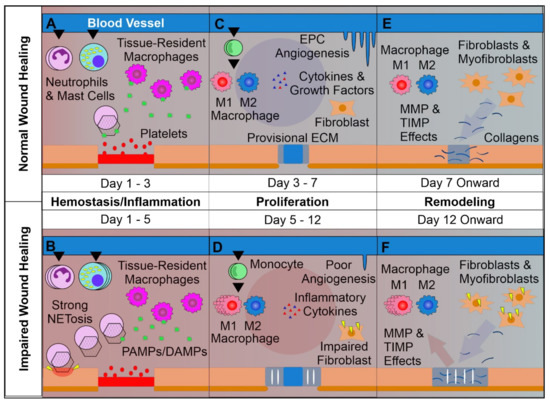
Figure 1. Overview of Normal vs. Impaired Wound Healing. (A): The first phase of wound healing is hemostasis. Platelets form a clot at the site of injury, and chemoattractants are released, recruiting key inflammatory cells. Next, inflammation takes charge, with infiltrating neutrophils and mast cells releasing pro-inflammatory cytokines and inducing strong sanitizing effects. This is accompanied by neutrophil extracellular trap (NETosis) induction, which assists in capturing and destroying invading pathogens. Tissue-resident macrophages react to pathogen- and damage-associated molecular patterns (PAMPs & DAMPs), activating. Later a provisional matrix comprised of fibronectin and other provisional extracellular matrix (ECM) components forms from the clot. (B): Impaired wounds see an upregulated influx of neutrophils and mast cells, leading to an overactive inflammatory response, causing collateral damage and extending the inflammatory phase to the detriment of subsequent phases. (C): Following resolution of strong inflammation, the proliferative phase begins. Crucially, endothelial progenitor cells are stimulated by growth factors to induce angiogenesis. This angiogenesis allows for wound-resident cells to be supplied with oxygen and nutrients, facilitating their function. Infiltrating monocytes differentiate into M1 and M2 macrophage subsets. M1 macrophages maintain a strong inflammatory profile, but are counterbalanced by pro-regenerative M2 macrophages which release anti-inflammatory cytokines, growth factors, and proteases which replace the provisional ECM with collagens, assisted by properly functioning fibroblasts. This process results in thick granular tissue and full keratinocyte coverage. (D): Impaired wounds result in poor angiogenesis and, in the case of T2DM, glycated proteins. This hypoxic environment induces oxidative stress, driving inflammatory M1 macrophage polarization and impairment of fibroblasts, resulting in poor ECM reorganization and a persistent inflammatory environment. (E): Remodeling is carried out by macrophages, fibroblasts, and myofibroblasts re-organizing the provisional ECM into a coherent scar structure primarily by means of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs), resulting in tissue with strong tensile strength and functionality. (F): Impaired wound-resident cells remain ineffective and pro-inflammatory. Collagen reorganization resolves poorly, resulting in weak, non-functional skin that is apt to re-injure and potentially ulcerate, perpetually inflamed.
It is widely recognized that localized, properly controlled inflammation acts as a trigger for the proliferative and remodeling phases [10][11]. On the other hand, the uncontrolled or prolonged inflammatory responses frequently observed in diabetic wounds lead to the impairment of subsequent phases of the wound healing process, or are implicated as a contributor to ulceration [12][13]. Local inflammation is strictly associated with neutrophil infiltration and activation. Interestingly, neutrophils are absent in unwounded skin and their trafficking to the wound area is induced and controlled by tissue-resident T cells, mast cells, and macrophages [14]. In fact, neutrophils represent an important source of proteases (including elastase, cathepsin G, and urokinase-type plasminogen activator—that support re-epithelialization) [15][16][17]; reactive oxygen and nitrogen species, cytokines (including IL-1β, tumor necrosis factor (TNF), IL-6, IL-12p40, and transforming growth factor β (TGF-β)), and chemokines (including CCL2, CCL3, CCL5, CXCL1, and CXCL2) [18]. Moreover, generally high neutrophil counts within the wound, and the consequently increased neutrophil-to-lymphocyte ratio is recognized as a hallmark of impaired wound healing observed in T2DM-affected individuals [19]. Interestingly, T2DM is known to induce neutrophil extracellular trap induction (NETosis), a phenomenon which may be responsible for delayed wound healing, given that disruption of neutrophil ability to undergo NETosis led to accelerated wound closure in previous studies (Figure 1) [12][20]. This continual activation of neutrophils and induction of NETosis results in the induction of yet more inflammation by way of mitochondrial DNA and histone H4 [21] in contrast with the normal process of inflammatory resolution by way of neutrophilic apoptotic body phagocytosis [11]. This uptake of neutrophil-derived apoptotic bodies’ by infiltrating monocytes/macrophages helps resolve the inflammatory phase in a self-perpetuating manner by limiting inflammatory cell infiltration and shifting the production of eicosanoids from pro-inflammatory to anti-inflammatory mediators [22][23][24]. Unfortunately, however, in diabetic wounds the inflammatory phase is significantly prolonged by the disruption of mechanisms which both control the influx of neutrophils as well as regulate their inflammatory processes [12][21][25]. Interestingly, it seems that the cause of many observed dysregulations of the inflammatory phase is not directly associated with localized high glucose levels but rather the epigenetic polarization of innate immune cell pro-inflammatory function prior to wound infiltration, as in progenitor cell modification due to T2DM-related systemic complications such as hyperglycemia [26]. This polarization of innate immune cells towards pro-inflammatory phenotypes is additionally supported by systemic inflammatory effects observed in diabetic patients and animal models [27]. However, to date, mechanisms controlling the epigenetic regulation of neutrophils and monocytes/macrophages in diabetic individuals remain elusive.
Monocyte migration to injured skin is controlled by chemokines derived by mast cells, keratinocytes, and fibroblasts acting through CCR2, CCR5, and mainly monocyte chemotactic protein 1 (MCP-1) [28]. Notably, due to pleiotropic biological activities, monocytes and macrophages are recognized as central players in the resolution and regulation of the inflammatory, proliferative, and remodeling phases of wound healing [29]. Unlike normal wound-infiltrating macrophages differentiating into classically-activated (inflammatory) M1 and alternatively-activated (reparative/regulatory) M2 macrophages, T2DM-affected macrophages strongly polarize into the inflammatory M1 phenotype [26]. Classically activated macrophages possess high phagocytic properties and are efficient in their production of pro-inflammatory cytokines—namely IL-1α, IL-1β, IL-6, TNF, and IL-12—contributing to and extending the inflammation phase, increasing neutrophilic infiltration, and prolonging low-grade inflammation which is characteristic of chronic DFUs [26][30]. Consequently, a lower absolute number of M2 macrophages and a higher M1:M2 macrophage ratio within the wound reduces secretory levels of growth factors PDGF, FGF, and VEGF, as well as anti-inflammatory cytokines including IL-10, TGF-α and TGF-β—all of which are responsible for the induction of the proliferative phase and effective regulation of inflammation, respectively [13][31]. Moreover, monocytes/macrophages act as antigen-presenting cells, linking the innate and adaptive immune responses [29]. Despite this, to date, the role of mutual interactions between macrophages and T cells in wound healing has yet to be fully elucidated.
It is well established that skin resident T cells play an essential role in the maintenance and regulation of local skin inflammation in the course of wound healing. In fact, Th17 cells were shown to promote neutrophilic infiltration, and high levels of IL-17A were shown to reduce wound repair [32]. On the other hand, regulatory T cells (Tregs) are considered essential regulators of inflammation and constitute a significant source of IL-10 [33]. Importantly, depletion of Tregs significantly reduces wound closure [34]. It is tempting to speculate that the systemic inflammation observed in diabetic patients limits the migration of Tregs and increases the infiltration of Th17 cells in the diabetic wound and thus represents one of the mechanisms of increased neutrophilic inflammation and a prolonged inflammatory phase. Notably, the healing process of diabetic wounds may be accelerated by topical retinoic acid, thereby inducing T cell plasticity and differentiation of Th17 cells towards Tregs [35]. This confirms the crucial role of T cells in the regulation of the inflammatory phase of diabetic wound healing.
Taken together, the prolonged inflammation phase observed in diabetic wounds that impairs wound closure and remodeling originates not only from high levels of localized pro-inflammatory mediators, but also from deficiencies in anti-inflammatory cytokines derived by regulatory cells, including M2 macrophages and Tregs. Despite this observation, the regulation of this process needs attention in future research.
References
- Cavan, D.; Fernandez, J.D.R.; Huang, Y.; Makaroff, L. IDF releases report of global survey on access to medicines and supplies for people with diabetes. Diabetes Res. Clin. Pract. 2017, 129, 224–225.
- Schwarz, P.E.; Gallein, G.; Ebermann, D.; Müller, A.; Lindner, A.; Rothe, U.; Nebel, I.T.; Müller, G. Global Diabetes Survey: An annual report on quality of diabetes care. Diabetes Res. Clin. Pract. 2013, 100, 11–18.
- Reiber, G.E.; McDonell, M.B.; Schleyer, A.M.; Fihn, S.D.; Reda, M.J. A comprehensive system for quality improvement in ambulatory care: Assessing the quality of diabetes care. Patient Educ. Couns. 1995, 26, 337–341.
- Baltzis, D.; Eleftheriadou, I.; Veves, A. Pathogenesis and Treatment of Impaired Wound Healing in Diabetes Mellitus: New Insights. Adv. Ther. 2014, 31, 817–836.
- Periayah, M.H.; Halim, A.S.; Saad, A.Z.M. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327.
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217.
- Gale, A.J. Continuing education course #2: Current understanding of hemostasis. Toxicol. Pathol. 2011, 39, 273–280.
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43.
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019.
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885.
- Peiseler, M.; Kubes, P. More friend than foe: The emerging role of neutrophils in tissue repair. J. Clin. Investig. 2019, 129, 2629–2639.
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819.
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030.
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Yager, D.R.; Nwomeh, B.C. The proteolytic environment of chronic wounds. Wound Repair Regen. 1999, 7, 433–441.
- Trengove, N.J.; Stacey, M.C.; Macauley, S.; Bennett, N.; Gibson, J.; Burslem, F.; Murphy, G.; Schultz, G. Analysis of the acute and chronic wound environments: The role of proteases and their inhibitors. Wound Repair Regen. 1999, 7, 442–452.
- Rømer, J.; Bugge, T.H.; Pyke, C.; Lund, L.R.; Flick, M.J.; Degen, J.L.; Danø, K.; Oslash, R.J. Impaired wound healing in mice with a disrupted plasminogen gene. Nat. Med. 1996, 2, 287–292.
- Etecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-Derived Cytokines: Facts beyond Expression. Front. Immunol. 2014, 5, 508.
- Vatankhah, N.; Jahangiri, Y.; Landry, G.J.; McLafferty, R.B.; Alkayed, N.J.; Moneta, G.L.; Azarbal, A.F. Predictive value of neutrophil-to-lymphocyte ratio in diabetic wound healing. J. Vasc. Surg. 2017, 65, 478–483.
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2019, 9, 3076.
- Brostjan, C.; Oehler, R. The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov. 2020, 6, 1–8.
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361.
- Widgerow, A.D. Cellular resolution of inflammation-catabasis. Wound Repair Regen. 2012, 20, 2–7.
- Freire-De-Lima, C.G.; Xiao, Y.Q.; Gardai, S.J.; Bratton, D.L.; Schiemann, W.P.; Henson, P.M. Apoptotic Cells, through Transforming Growth Factor-β, Coordinately Induce Anti-inflammatory and Suppress Pro-inflammatory Eicosanoid and NO Synthesis in Murine Macrophages. J. Biol. Chem. 2006, 281, 38376–38384.
- Yang, S.; Gu, Z.; Lu, C.; Zhang, T.; Guo, X.; Xue, G.; Zhang, L. Neutrophil Extracellular Traps Are Markers of Wound Healing Impairment in Patients with Diabetic Foot Ulcers Treated in a Multidisciplinary Setting. Adv. Wound Care 2020, 9, 16–27.
- Yan, J.; Tie, G.; Wang, S.; Tutto, A.; Demarco, N.; Khair, L.; Fazzio, T.G.; Messina, L.M. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat. Commun. 2018, 9, 1–13.
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119.
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interf. Cytokine Res. 2009, 29, 313–326.
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23.
- Tellechea, A.; Leal, E.C.; Kafanas, A.; Auster, M.E.; Kuchibhotla, S.; Ostrovsky, Y.; Tecilazich, F.; Baltzis, D.; Zheng, Y.; Carvalho, E.; et al. Mast Cells Regulate Wound Healing in Diabetes. Diabetes 2016, 65, 2006–2019.
- Gonzalez, A.C.D.O.; Andrade, Z.D.A.; Costa, T.F.; Medrado, A.R.A.P. Wound Healing: A Literature Review. An. Bras. Dermatol. 2016, 91, 614–620.
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Constantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343.
- O’Garra, A.; Vieira, P.L.; Vieira, P.; Goldfeld, A.E. IL-10-producing and naturally occurring CD4+ Tregs: Limiting collateral damage. J. Clin. Investig. 2004, 114, 1372–1378.
- Nosbaum, A.; Prevel, N.; Truong, H.A.; Mehta, P.; Ettinger, M.; Scharschmidt, T.C.; Ali, N.H.; Pauli, M.L.; Abbas, A.K.; Rosenblum, M.D. Cutting Edge: Regulatory T Cells Facilitate Cutaneous Wound Healing. J. Immunol. 2016, 196, 2010–2014.
- Van, Y.H.; Lee, W.H.; Ortiz, S.; Lee, M.H.; Qin, H.J.; Liu, C.P. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-gamma-producing T-cells without affecting Th17 cells. Diabetes 2009, 58, 146–155.


