 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dirk Reinhardt | + 2052 word(s) | 2052 | 2021-03-23 11:14:50 | | | |
| 2 | Rita Xu | Meta information modification | 2052 | 2021-04-01 04:55:52 | | |
Video Upload Options
Acute myeloid leukemia is a life-threatening malignant disorder arising in a complex and dysregulated microenvironment that, in part, promotes the leukemogenesis. Treatment of relapsed and refractory AML, despite the current overall success rates in management of pediatric AML, remains a challenge with limited options considering the heavy but unsuccessful pretreatments in these patients.
1. Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy that originates from transformed myeloid precursor cells arising from a hijacked bone marrow microenvironment (BMM). Leukemogenesis is characterized by uncontrolled clonal proliferation of malignant leukemic cells (blasts) that have lost the ability of proper differentiation at various stages of maturation. Our knowledge today suggests that leukemic blasts transduce the surrounding BMM into a leukemia-supportive niche and vice-versa, pointing at a bidirectional crosstalk between leukemic blasts and BMM reciprocally supporting further disease progression In adults, AML represents the most common form of acute leukemia whilst in pediatrics, it accounts for 20% of all childhood leukemias with an overall survival of about 70% that ranges from 60% to 90% depending on the risk profile [1][2]. However, the prognosis is still poor in cases of refractory disease and relapse, which occur in about 30% of the patients [3][4].
Considering its heterogeneous characteristics, treatment of pediatric AML is adapted to different risk groups, stratified based on different genetic, cytogenetic, and clinical properties. Primarily though, treatment in all groups consists of intensive chemotherapeutic regimens with severe systemic side effects, emphasizing the urgent need for more tolerable, less toxic, and highly efficient treatments. Stepping towards this goal, numerous research works have uncovered substantial mechanisms underlying leukemogenesis and provided pivotal knowledge regarding the biology of AML, paving the way for identification of promising novel therapeutic approaches [5]. However, some of the targeted therapeutic attempts failed to approve desired efficiency and safety in early phase trials and only a few have entered the clinic (examples regarding antibody-based immunotherapies[6][7][7][8][9] and regarding immune-checkpoint-inhibitor therapies [10][11][12]). Encouraged by the graft-versus-leukemia (GvL) effect following allogenic hematopoietic stem cell transplantation (HSCT) in liquid cancers and the reported success of immunotherapy in solid tumors, immunological treatment opportunities have gradually gained attention [13][14]. Allo-HSCT is one of the oldest and best-known immunotherapies for AML. It has been proven capable of eradicating the residual disease and preventing relapse after the failure of first-line treatment in high-risk patients. The efficacy of HSCT is, however, limited by the severe chemotherapy-related toxicities during conditioning, in acute or chronic graft-versus-host disease (GvHD), or in the event of relapse. Although AML is historically known as an immuno-responsive disease, leukemic blasts reside in a highly supportive, immunosuppressive environment where they adopt various strategies to evade immune surveillance. To date, major efforts have been made to develop new ways to uncover hidden leukemic blasts and to restore intrinsic anti-leukemic immuno-surveillance. Concerning the impact of BMM, which has been shown to be immunosuppressive in AML, successful immunotherapy should target both the immunologically dysregulated microenvironment and the malignant blasts that can escape the immune surveillance.
2. Acute Myeloid Leukemia Harnesses the Immunological Microenvironment
In addition to oncogenic alterations in hematopoietic cells and BMM, immunological dysregulations contribute to leukemogenesis as well. During the leukemic transition, leukemic stem cells undergo immunoediting, a process that comprises the acquisition of multiple strategies to successfully evade immune surveillance. Consequently, the selected leukemic population is characterized by different immune-evasive mechanisms (Figure 1).
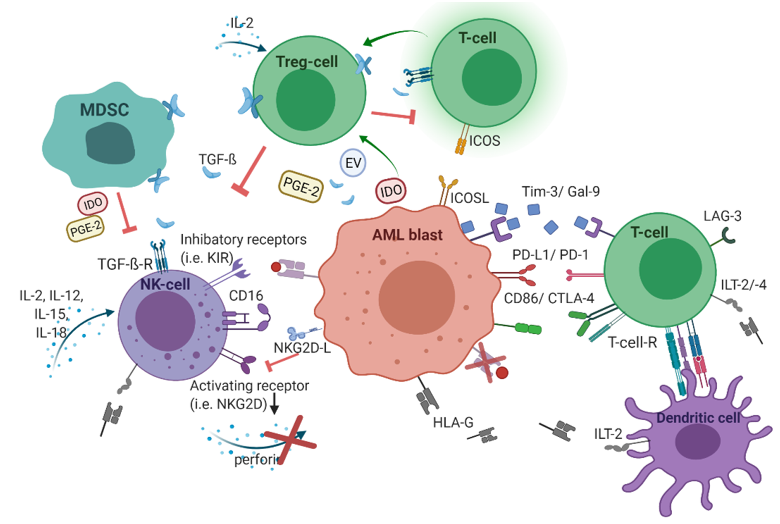
Figure 1. The immunological microenvironment in acute myeloid leukemia (AML). AML blasts reduce antigen-presentation through downregulation of classical human-leukocyte-antigen (HLA)-presentation. Non-classical HLA-G is supposed to suppress immunogenicity. Checkpoint molecules promote immune evasion (Gal-9/Tim-3, PD-L1/PD-1, CD86/CTLA-4, and LAG-3). Secretion of TGF-β and indoleamine 2,3-dioxygenase (IDO), as well as inducible T-cell-co-stimulator ligand (ICOS)/ICOS-ligand interplay, induces T-cell conversion into immunosuppressive T-regulatory cells (Treg) cells. Myeloid-derived suppressor cells (MDSC) suppress natural killer (NK)-cell-mediated cytotoxicity, i.e., via IDO, prostaglandin-E2, and TGF-β. Figure 1 was created with biorender.com (accessed on 20 February 2021).
2.1. Human Leukocyte Antigens
One known mechanism of immune tolerance in AML was initially observed in patients with relapsed AML following mismatched HSCT, i.e., partially human-leukocyte-antigen (HLA)-incompatible HSCT, which contributes to downregulation of the mismatched HLA class I and II in AML blasts. Upon pressure from the transplanted immune system, this strategy confers ‘survival advantage’ to outgrowing immune-resistant mutant AML clones, characterized by genomic loss of the mismatched histocompatibility determinants. Leukemic blasts evade alloreactive donor T-cell-recognition and killing through this genomic loss of mismatched HLA haplotype which hampers the GvL effect following allogeneic HSCT; thus paving the way for leukemia relapse [15][16][17][18][19].
The non-classical HLA class I molecule HLA-G physiologically suppresses the immune system by direct inhibition of dendritic cells (via the inhibitory receptors immunoglobulin-like transcript (ILT)-2 and ILT-4), T-cells (via ILT-2), natural killer (NK)-cells (via ILT-2 and the killer-immunoglobulin-like-receptor (KIR)-2DL4), and monocytes (via ILT-2) [20]. HLA-G has been explored in multiple cancers and its biological and clinical impact as a possible checkpoint inhibitor has been carefully studied [21]. It has been shown that the soluble isoforms of HLA-G are increased in distinct AML samples, especially in monocytic lineages, following interferon (IFN)-gamma and Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) stimulation [22][23]. In addition, different isoforms of HLA-G have been reported to be secreted or expressed by leukemic cells associated with a higher blast percentage in bone marrow, decreased T cell number, and relapses, which may indicate HLA-G as an additional strategy for AML blasts to evade immune surveillance [24][25]. However, the clinical impact of HLA-G in leukemogenesis appears to be controversial as opposing results do not confirm the clinical implications of HLA-G [26]. In particular, one report did not detect HLA-G in leukemic samples [27].
2.2. Checkpoint Molecules
To evade immune surveillance, leukemic blasts (relapse/refractory rather than newly diagnosed patients) express programmed-cell-death ligand-1 (PD-L1), a checkpoint marker that compromises cytotoxic T cells expressing PD-1 [28]. Blockade of PD-L1/PD-1 restores functionality to the exhausted cytotoxic T-cells while synergistically debilitating Treg-mediated immunosuppression, which leads to decreased tumor-burden in an adoptively transferred AML-murine model [29]. Additionally, inhibition of the co-expressed checkpoint-marker T-cells immunoglobulin-mucin 3 (Tim-3) on leukemic blasts generated an even stronger anti-leukemic effect in an AML-murine model [31]. Binding between Tim-3 on leukemic cells and its ligand galectin-9, which is highly expressed in AML blasts, promotes self-renewal via stimulatory β-catenin and NFkB-signaling and reduces the release of pro-inflammatory cytokines resulting in NK- and T-cell dysfunction [31][32]. In addition, the inhibitory checkpoint-marker C-type lectin-like inhibitory-receptor (CTLA)-4, that competes with CD28, binding CD80/CD86 on leukemic blasts and lymphocyte-activating gene (LAG)-3 has been detected upregulated in primary AML samples. These markers contribute to poor outcome, especially when concurrently expressed in patterns with PD-L1 and/or PD-L2 on leukemic cells [30][33].
2.3. T-cellular Immune Dysregulation
In the leukemic microenvironment, T-cells are proposed to reveal altered functional and phenotypic profiles, thereby contributing to immune-suppressive surroundings [34][35][36]. Consistently, the proportion of inhibitory CD4+CD25+ T-regulatory (Treg) cells has been found markedly increased in AML-patient-derived peripheral blood samples and these Treg cells were shown to reduce T-cell proliferation and cytokine production, i.e., IFN-gamma and interleukin (IL)-2 [29][37][38]. In the context of T-cell immunity, the cytokine IL-2 has been proposed to exert two-sided regulative effects. On the one hand, IL-2 supports leukemia-directed lymphocytes, on the other, IL-2 simultaneously favors Treg cells driving the immune-suppressive leukemic microenvironment [39][40]. It has been concluded that besides cell-to-cell contacts, secretion of the immunoinhibitory factors IL-10 and transforming growth factor-beta (TGF-β) contribute to T-reg-mediated suppression of T-cell proliferation [38]. Notably, leukemic cells expressing high indoleamine 2,3-dioxygenase (IDO) were associated with elevated Treg cells [41], probably because of the finding that IDOs induce T-cell conversion into suppressive Treg cells [42]. In addition, AML cells overexpressing the inducible T-cell-co-stimulator ligand (ICOSL) also evoke T-cell conversion, driving the expansion of Treg cells that secrete increased levels of IL-10 and, thereby, favor proliferation and stemness in AML blasts [43]. Hence, AML cells themselves apparently engender T-cell tolerance; thus promoting leukemia progression. In addition, the immunomodulating mediator TGF-β also converts T-cells into Treg cells [44].
Apart from Treg-cell-mediated T-cell suppression, leukemic cells have also been proposed to orchestrate arginase and STAT-3 pathways to reduce T-cell proliferation, which was restored after selective inhibition, confirming the immune-suppressive impact of these mediators [34][37]. Inhibition of cytokine production and cell cycle entry of T-cells exposed to leukemic cell-derived supernatant has been explained by secreted proteins affecting cellular pathways [45]. Intriguingly, previous reports analyzing the effect of AML-cell supernatant on stimulated lymphocytes revealed that cytotoxicity was not affected even though the proliferative capacity of exposed T-cells was inhibited [46]. Schnorfeil et al. observed neither inhibition of T-cell proliferation nor activity in AML samples at different stages of disease and, consequently, reasoned that T-cell-based immunotherapies may have favorable prospects in AML [47]. More specifically, leukemia antigen-directed T-cell responses have been suggested to be increased when the leukemia burden is minimal due to the investigation that immunocompetent mice transplanted with MLL/AF9-leukemia showed a spontaneous antigen-specific T-cell response when a minimum number of leukemia-initiating cells were injected, whereas T-cell immunity was exhausted in mice with advanced leukemia [48].
In conclusion, as suggested by numerous research works [34][35][36][45][46][49][50][51], T-cell immunity appears to be compromised in the leukemic microenvironment and an increased proportion of inhibitory Treg cells importantly contribute to this. Nevertheless, data are still limited and exact functional status, as well as underlying mechanisms, have largely remained obscure.
2.4. NK Cell-Related Strategies of Immune Evasion
Multiple studies provide evidence of deregulated anti-leukemic NK-mediated cytotoxicity in AML [52][53][54][55][56][57][58]. Leukemic blasts have been revealed to reduce NK activity by different mechanisms. Sera samples taken from AML patients were enriched in microvesicles containing increased levels of TGF-β, which led to reduced anti-leukemic NK-cytotoxicity in vitro [52]. Conversely, in an NK cell—AML-blasts co-culture system, Stringaris et al. detected an increased level of IL-10 but not TGF-β, suggesting that inhibition of NK cells by AML blasts depends on IL-10 instead [57]. Myeloid-derived suppressor cells (MDSCs) are a distinct immature cell population that facilitates immune-evasive strategies [59]. Recently, MDSCs were shown to suppress the anti-leukemic activity of NK cells, mediated by IDO and prostaglandin-E2 (PGE2) and exosomes [60]. AML blasts are supposed to constitutively express IDO, nonetheless, the exact contribution to AML progression and immune tolerance remains to be elucidated [61]. Leukemic blasts induce MDSC proliferation and differentiation into tumor-associated macrophages, which further inhibit immunogenicity favoring their survival; thus driving leukemia progression resulting in deteriorated outcomes [59][62][63]. Deficient NK cell function can be traced back to the weak expression of different activating natural cytotoxic receptors (NCRs) on AML-derived NK cells [53][54][55]. Since a small number of AML-derived NK cell samples showed diminished anti-leukemic activity regardless of expressing high NCR-levels, it is suggested that in some cases, AML cells express a low level of NCR ligands to escape NK-mediated cytotoxicity [54][56]. NK- and T-cells also recognize cancer cells through the activating receptor NK-activating surface marker (NKG2D)-binding to its ligands MHC class I-related chain (MIC) -A and -B and UL16-binding protein (ULBP)-1 and -2 [64]. In acute leukemia, the expression of NKG2D is downregulated or not present at all [57,66]. Leukemic cells contribute to this immune-evading strategy since they release increased levels of TGF-β [52][66]. Moreover, secretion of soluble NKG2D-ligands by leukemic blasts inhibits NGK2D expression on NK cells, resulting in decreased anti-leukemia activity [67]. Of note, stem-cell-like subsets of AML blasts that lack surface-bound NKG2D-ligands (attributed to elevated poly-ADP-ribose-polymerase-1 (PARP1)) efficiently evade NK-driven immune control, conferring a selective advantage in the absence of NKG2D ligands [68]. Impaired binding of perforin is another resistance mechanism through which distinct AML blasts can elude perforin-mediated NK cell-lysis [69]. Mesenchymal stem cells decrease NK-cytotoxicity and IL-2-induced NK cell expansion by secretion of IDO and the cyclooxygenase (COX)-2 product, prostaglandin E2 (PGE2) [70]. In addition, PGE2 has been revealed to suppress IL-15-stimulated NK cell reactivity, in terms of cytotoxicity and IFN-gamma egress [71]. Furthermore, proliferation and differentiation of CD8+ T-cells are reported to be inhibited by diminished tryptophan levels, owing to enzymatic digestion by IDO [72] and its catabolite was shown to decrease NK-activating surface marker (NKG2D) [73]. Also, the enzymatic activity of IDO, highly expressed in BM stromal cells causes the conversion into Treg cells [42][74] and boosts their immune-suppressive function [75][76]. In addition to the before-mentioned Treg-cell-mediated T-cell suppression, Treg cells have been explored to actively reduce NK cell cytotoxicity and NKG2D receptor expression through membrane-bound TGF-β [77]. These results may imply the high value of TGF-β, PGE2, and IDO disturbing the immunogenic anticancer response. However, more research is required to explore the decisive impact of immunological dysregulating factors in the BMM, especially in AML.
References
- Ladikou, E.E.; Sivaloganathan, H.; Pepper, A.; Chevassut, T. Acute Myeloid Leukaemia in Its Niche: the Bone Marrow Microenvironment in Acute Myeloid Leukaemia. Curr. Oncol. Rep. 2020, 22, doi:10.1007/s11912-020-0885-0.
- Mareike Rasche; Martin Zimmermann; Lisa Borschel; Jean-Pierre Bourquin; Michael Dworzak; Thomas Klingebiel; Thomas Lehrnbecher; Ursula Creutzig; Jan-Henning Klusmann; Dirk Reinhardt. Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177, doi:10.1038/s41375-018-0071-7.
- Rasche, M.; Steidel, E.; Kondryn, D.; Neuhoff, N. von; Sramkova, L.; Creutzig, U.; Dworzak, M.; Reinhardt, D. Impact of a Risk-Adapted Treatment Approach in Pediatric AML: A Report of the AML-BFM Registry 2012. Blood 2019, 134, 293, doi:10.1182/blood-2019-130969.
- Li, S.; Mason, C.E.; Melnick, A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr. Opin. Genet. Dev. 2016, 36, 100–106, doi:10.1016/j.gde.2016.03.011.
- Davila, J.; Slotkin, E.; Renaud, T. Relapsed and refractory pediatric acute myeloid leukemia: current and emerging treatments. Paediatr. Drugs 2014, 16, 151–168, doi:10.1007/s40272-013-0048-y.
- Winer, E.S.; Stone, R.M. Novel therapy in Acute myeloid leukemia (AML): moving toward targeted approaches. Ther. Adv. Hematol. 2019, 10, 2040620719860645, doi:10.1177/2040620719860645.
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 4854–4860, doi:10.1182/blood-2013-01-466706.
- He, S.Z.; Busfield, S.; Ritchie, D.S.; Hertzberg, M.S.; Durrant, S.; Lewis, I.D.; Marlton, P.; McLachlan, A.J.; Kerridge, I.; Bradstock, K.F.; et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leukemia & lymphoma 2015, 56, doi:10.3109/10428194.2014.956316.
- Feldman, E.J.; Brandwein, J.; Stone, R.; Kalaycio, M.; Moore, J.; O'Connor, J.; Wedel, N.; Roboz, G.J.; Miller, C.; Cho-pra, R.; et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lin-tuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J. Clin. Oncol. 2005, 23, doi:10.1200/JCO.2005.09.133.
- Morsink, L.M.; Walter, R.B. Novel monoclonal antibody-based therapies for acute myeloid leukemia. Best Practice & Research Clinical Haematology 2019, 32, 116–126, doi:10.1016/j.beha.2019.05.002.
- Albring, J.C.; Inselmann, S.; Sauer, T.; Schliemann, C.; Altvater, B.; Kailayangiri, S.; Rössig, C.; Hartmann, W.; Knor-renschild, J.R.; Sohlbach, K.; et al. PD-1 checkpoint blockade in patients with relapsed AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2017, 317–320, doi:10.1038/bmt.2016.274.
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoiet-ic cell transplantation. Blood 2009, 113, 1581–1588, doi:10.1182/blood-2008-07-168468.
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153, doi:10.1056/NEJMoa1601202.
- Stephan Kruger; Matthias Ilmer; Sebastian Kobold; Bruno L. Cadilha; Stefan Endres; Steffen Ormanns; Gesa Schuebbe; Bernhard W. Renz; Jan G. D’Haese; Hans Schloesser; et al. Advances in cancer immunotherapy 2019 – latest trends. J Exp Clin Cancer Res 2019, 38, 1–11, doi:10.1186/s13046-019-1266-0.
- Sweeney, C.; Vyas, P. The Graft-Versus-Leukemia Effect in AML. Front. Oncol. 2019, 9, doi:10.3389/fonc.2019.01217.
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Invest. 2020, 130, 1552–1564, doi:10.1172/JCI129204.
- Stölzel, F.; Hackmann, K.; Kuithan, F.; Mohr, B.; Füssel, M.; Oelschlägel, U.; Thiede, C.; Röllig, C.; Platzbecker, U.; Schetelig, J.; et al. Clonal evolution including partial loss of human leukocyte antigen genes favoring extramedul-lary acute myeloid leukemia relapse after matched related allogeneic hematopoietic stem cell transplantation. Transplantation 2012, 93, 744–749, doi:10.1097/TP.0b013e3182481113.
- Toffalori, C.; Riba, M.; Zito, L.; Barcella, M.; Spinelli, O.; Crucitti, L.; Cieri, N.; Peccatori, J.; Bernardi, M.; Bonini, C.; et al. Acute Myeloid Leukemia Relapses after Allogenenic HSCT Display a Distinctive Immune-Related Signature, with Frequent and Functionally Relevant Alterations in HLA Class II Antigen Presentation and T Cell Costimula-tion. Blood 2014, 124, 427, doi:10.1182/blood.V124.21.427.427.
- Crucitti, L.; Crocchiolo, R.; Toffalori, C.; Mazzi, B.; Greco, R.; Signori, A.; Sizzano, F.; Chiesa, L.; Zino, E.; Lupo Stanghellini, M.T.; et al. Incidence, risk factors and clinical outcome of leukemia relapses with loss of the mis-matched HLA after partially incompatible hematopoietic stem cell transplantation. Leukemia 2015, 29, 1143–1152, doi:10.1038/leu.2014.314.
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O'Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341, doi:10.1056/NEJMoa1808777.
- Colonna, M.; Samaridis, J.; Cella, M.; Angman, L.; Allen, R.L.; O'Callaghan, C.A.; Dunbar, R.; Ogg, G.S.; Cerundolo, V.; Rolink, A. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J. Immunol. 1998, 160, 3096–3100.
- Lin, A.; Yan, W.-H. Human Leukocyte Antigen-G (HLA-G) Expression in Cancers: Roles in Immune Evasion, Me-tastasis and Target for Therapy. Mol. Med. 2015, 21, 782–791, doi:10.2119/molmed.2015.00083.
- Gros, F.; Sebti, Y.; Guibert, S. de; Branger, B.; Bernard, M.; Fauchet, R.; Amiot, L. Soluble HLA-G molecules increase during acute leukemia, especially in subtypes affecting monocytic and lymphoid lineages. Neoplasia 2006, 8, 223–230, doi:10.1593/neo.05703.
- Mizuno, S.; Emi, N.; Kasai, M.; Ishitani, A.; Saito, H. Aberrant expression of HLA-G antigen in interferon gam-ma-stimulated acute myelogenous leukaemia. Br. J. Haematol. 2000, 111, doi:10.1046/j.1365-2141.2000.02345.x.
- Yan, W.-H.; Lin, A.; Chen, B.-G.; Luo, W.-D.; Dai, M.-Z.; Chen, X.-J.; Xu, H.-H.; Li, B.-L. Unfavourable clinical impli-cations for HLA-G expression in acute myeloid leukaemia. J. Cell. Mol. Med. 2008, 12, 889–898, doi:10.1111/j.1582-4934.2008.00175.x.
- Hamed, N.M.; El Halawani, N.; Nafea, D.; El Rahman, M.; Kasber, A. Soluble HLA-G: A novel marker in acute my-eloid leukemia patients. Acta Med Int 2017, 4, 51, doi:10.5530/ami.2017.4.10.
- Guo, Q.Y.; Chen, B.G.; Ruan, Y.Y.; Lin, A.; Yan, W.H. HLA-G expression is irrelevant to prognosis in patients with acute myeloid leukemia. Leukemia research 2011, 35, 1350–1354, doi:10.1016/j.leukres.2011.05.036.
- Poláková, K.; Krcová, M.; Kuba, D.; Russ, G. Analysis of HLA-G expression in malignant hematopoetic cells from leukemia patients. Leukemia research 2003, 27, doi:10.1016/s0145-2126(02)00228-x.
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood 2009, 114, 1545–1552, doi:10.1182/blood-2009-03-206672.
- Zhou, Q.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Lev-ine, B.L.; et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010, 116, 2484–2493, doi:10.1182/blood-2010-03-275446.
- Zhou, Q.; Munger, M.E.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Ander-son, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011, 117, 4501–4510, doi:10.1182/blood-2010-10-310425.
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.-i.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Mye-loid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell 2015, 17, 341–352, doi:10.1016/j.stem.2015.07.011.
- Gonçalves, S.I.; Im Yasinska; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Sili-gardi, G.; Ceccone, G.; et al. The Tim-3-galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine 2017, 22, doi:10.1016/j.ebiom.2017.07.018.
- Chen, C.; Liang, C.; Wang, S.; Chio, C.L.; Zhang, Y.; Zeng, C.; Chen, S.; Wang, C.; Li, Y. Expression patterns of im-mune checkpoints in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 28, doi:10.1186/s13045-020-00853-x.
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, doi:10.1172/jci.insight.120974.
- Li, Z.; Philip, M.; Ferrell, P.B. Alterations of T-cell-mediated immunity in acute myeloid leukemia. Oncogene 2020, 39, 3611–3619, doi:10.1038/s41388-020-1239-y.
- Mussai, F.; Santo, C. de; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758, doi:10.1182/blood-2013-01-480129.
- Wang, X.; Zheng, J.; Liu, J.; Yao, J.; He, Y.; Li, X.; Yu, J.; Yang, J.; Liu, Z.; Huang, S. Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur. J. Haematol. 2005, 75, 468–476, doi:10.1111/j.1600-0609.2005.00537.x.
- Szczepanski, M.J.; Szajnik, M.; Czystowska, M.; Mandapathil, M.; Strauss, L.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leu-kemia. Clin. Cancer Res. 2009, 15, 3325–3332, doi:10.1158/1078-0432.CCR-08-3010.
- Liao, W.; Lin, J.-X.; Leonard, W.J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunother-apy. Immunity 2013, 38, 13–25, doi:10.1016/j.immuni.2013.01.004.
- Ettinghausen, S.E.; Lipford, E.H.; Mulé, J.J.; Rosenberg, S.A. Recombinant interleukin 2 stimulates in vivo prolifera-tion of adoptively transferred lymphokine-activated killer (LAK) cells. The Journal of Immunology 1985, 135, 3623.
- Arandi, N.; Ramzi, M.; Safaei, F.; Monabati, A. Overexpression of indoleamine 2,3-dioxygenase correlates with reg-ulatory T cell phenotype in acute myeloid leukemia patients with normal karyotype. Blood Res. 2018, 53, 294–298, doi:10.5045/br.2018.53.4.294.
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877, doi:10.1182/blood-2006-07-036863.
- Han, Y.; Dong, Y.; Yang, Q.; Xu, W.; Jiang, S.; Yu, Z.; Yu, K.; Zhang, S. Acute Myeloid Leukemia Cells Express ICOS Ligand to Promote the Expansion of Regulatory T Cells. Front. Immunol. 2018, 9, 2227, doi:10.3389/fimmu.2018.02227.
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. The Journal of experimental medicine 2003, 198, doi:10.1084/jem.20030152.
- Buggins, A.G.; Milojkovic, D.; Arno, M.J.; Lea, N.C.; Mufti, G.J.; Thomas, N.S.; Hirst, W.J. Microenvironment pro-duced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J. Immunol. 2001, 167, 6021–6030, doi:10.4049/jimmunol.167.10.6021.
- Orleans-Lindsay, J.K.; Barber, L.D.; Prentice, H.G.; Lowdell, M.W. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function--implications for the adoptive immuno-therapy of leukaemia. Clin. Exp. Immunol. 2001, 126, 403–411, doi:10.1046/j.1365-2249.2001.01692.x.
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T cells are functionally not impaired in AML: increased PD-1 expression is only seen at time of relapse and cor-relates with a shift towards the memory T cell compartment. J. Hematol. Oncol. 2015, 8, doi:10.1186/s13045-015-0189-2.
- Hasegawa, K.; Tanaka, S.; Fujiki, F.; Morimoto, S.; Nakajima, H.; Tatsumi, N.; Nakata, J.; Takashima, S.; Nishida, S.; Tsuboi, A.; et al. An Immunocompetent Mouse Model for MLL/AF9 Leukemia Reveals the Potential of Spontaneous Cytotoxic T-Cell Response to an Antigen Expressed in Leukemia Cells. PLoS One 2015, 10, e0144594, doi:10.1371/journal.pone.0144594.
- Le Dieu, R.; Taussig, D.C.; Ramsay, A.G.; Mitter, R.; Miraki-Moud, F.; Fatah, R.; Lee, A.M.; Lister, T.A.; Gribben, J.G. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood 2009, 114, 3909–3916, doi:10.1182/blood-2009-02-206946.
- Jia, B.; Zhao, C.; Rakszawski, K.L.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Wang, M.; Shike, H.; Bayerl, M.G.; et al. Eomes+T-betlow CD8+ T Cells Are Functionally Impaired and Are Associated with Poor Clinical Outcome in Patients with Acute Myeloid Leukemia. Cancer Res. 2019, 79, 1635–1645, doi:10.1158/0008-5472.CAN-18-3107.
- van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progres-sion and Immunity. Cell 2019, 176, 1265-1281.e24, doi:10.1016/j.cell.2019.01.031.
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309, doi:10.3324/haematol.2010.039743.
- Fauriat, C.; Just-Landi, S.; Mallet, F.; Arnoulet, C.; Sainty, D.; Olive, D.; Costello, R.T. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007, 109, 323–330, doi:10.1182/blood-2005-08-027979.
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.-J.; Reviron, D.; Gastaut, J.-A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667, doi:10.1182/blood.v99.10.3661.
- Venton, G.; Labiad, Y.; Colle, J.; Fino, A.; Afridi, S.; Torres, M.; Monteuil, S.; Loriod, B.; Fernandez-Nunez, N.; Far-nault, L.; et al. Natural killer cells in acute myeloid leukemia patients: from phenotype to transcriptomic analysis. Immunol. Res. 2016, 64, 1225–1236, doi:10.1007/s12026-016-8848-0.
- Nowbakht, P.; Ionescu, M.-C.S.; Rohner, A.; Kalberer, C.P.; Rossy, E.; Mori, L.; Cosman, D.; Libero, G. de; Wodnar-Filipowicz, A. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood 2005, 105, 3615–3622, doi:10.1182/blood-2004-07-2585.
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; Lavallade, H. de; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847, doi:10.3324/haematol.2013.087536.
- Mundy-Bosse, B.; Kathleen, M.; Mao, C.; Ahmed, E.; Chen, L.; Scoville, S.D.; Freud, A.G.; Yu, J.; Caligiuri, M.A. Acute Myeloid Leukemia Alters Natural Killer Cell Maturation and Functional Activation. Blood 2014, 124, 754, doi:10.1182/blood.V124.21.754.754.
- Hyun, S.Y.; Na, E.J.; Jang, J.E.; Chung, H.; Kim, S.J.; Kim, J.S.; Kong, J.H.; Shim, K.Y.; Lee, J.I.; Min, Y.H.; et al. Immu-nosuppressive role of CD11b+ CD33+ HLA-DR- myeloid-derived suppressor cells-like blast subpopulation in acute myeloid leukemia. Cancer Med. 2020, doi:10.1002/cam4.3360.
- Tumino, N.; Besi, F.; Di Pace, A.L.; Mariotti, F.R.; Merli, P.; Li Pira, G.; Galaverna, F.; Pitisci, A.; Ingegnere, T.; Pelosi, A.; et al. PMN-MDSC are a new target to rescue graft-versus-leukemia activity of NK cells in haplo-HSC transplan-tation. Leukemia 2020, 34, 932–937, doi:10.1038/s41375-019-0585-7.
- Sobash, P.T.; Kolhe, R.; Karim, N.A.; Guddati, A.K.; Jillella, A.; Kota, V. Role of indoleamine 2,3-dioxygenase in acute myeloid leukemia. Future Oncol. 2020, doi:10.2217/fon-2019-0642.
- Gao, L.; Yu, S.; Zhang, X. Hypothesis: Tim-3/galectin-9, a new pathway for leukemia stem cells survival by pro-moting expansion of myeloid-derived suppressor cells and differentiating into tumor-associated macrophages. Cell Biochem. Biophys. 2014, 70, 273–277, doi:10.1007/s12013-014-9900-0.
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1801, doi:10.1182/blood-2016-07-730614.
- López-Larrea, C.; Suárez-Alvarez, B.; López-Soto, A.; López-Vázquez, A.; Gonzalez, S. The NKG2D receptor: sens-ing stressed cells. Trends in molecular medicine 2008, 14, doi:10.1016/j.molmed.2008.02.004.
- Pende, D.; Spaggiari, G.M.; Marcenaro, S.; Martini, S.; Rivera, P.; Capobianco, A.; Falco, M.; Lanino, E.; Pierri, I.; Zambello, R.; et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolat-ed myeloid or lymphoblastic leukemias: evidence for the involvement of the Poliovirus receptor (CD155) and Nec-tin-2 (CD112). Blood 2005, 105, 2066–2073, doi:10.1182/blood-2004-09-3548.
- Hong, C.-S.; Muller, L.; Whiteside, T.L.; Boyiadzis, M. Plasma exosomes as markers of therapeutic response in pa-tients with acute myeloid leukemia. Front. Immunol. 2014, 5, 160, doi:10.3389/fimmu.2014.00160.
- Hilpert, J.; Grosse-Hovest, L.; Grünebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Compre-hensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371, doi:10.4049/jimmunol.1200796.
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259, doi:10.1038/s41586-019-1410-1.
- Lehmann, C.; Zeis, M.; Schmitz, N.; Uharek, L. Impaired binding of perforin on the surface of tumor cells is a cause of target cell resistance against cytotoxic effector cells. Blood 2000, 96, 594–600.
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, doi:10.1182/blood-2007-02-074997.
- Joshi, P.C.; Zhou, X.; Cuchens, M.; Jones, Q. Prostaglandin E2 suppressed IL-15-mediated human NK cell function through down-regulation of common gamma-chain. J. Immunol. 2001, 166, 885–891, doi:10.4049/jimmunol.166.2.885.
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Sautès-Fridman, C.; Tartour, E.; Kennedy, E.P.; Platten, M.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2014, 3, e957994, doi:10.4161/21624011.2014.957994.
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating re-ceptors and regulates NK-cell function. Blood 2006, 108, 4118–4125, doi:10.1182/blood-2006-03-006700.
- Mansour, I.; Zayed, R.A.; Said, F.; Latif, L.A. Indoleamine 2,3-dioxygenase and regulatory T cells in acute myeloid leukemia. Hematology 2016, 21, 447–453, doi:10.1080/10245332.2015.1106814.
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.-Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 2007, 117, 2570–2582, doi:10.1172/JCI31911.
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207, doi:10.1016/j.it.2016.01.002.


