 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nuno Gonçalves-Anjo | + 2334 word(s) | 2334 | 2021-03-23 07:07:09 | | | |
| 2 | Vivi Li | Meta information modification | 2334 | 2021-03-24 05:24:54 | | |
Video Upload Options
Prion is defined as a “proteinaceous infectious particle” consisting exclusively of a single protein without the involvement of nucleic acids that causes spongiform encephalopathies in mammals. Prion diseases are characterized by the accumulation of abnormal isoforms of PrP glycoprotein.
1. Introduction
Even though the histopathological examination is a less sensitive diagnostic method for transmissible spongiform encephalopathies (TSEs) compared to detection by immunohistochemistry, PETblot and Western blot of PrPSc, the prion diagnostic marker, it is indispensable in the study of the lesions in TSEs to identify and characterize TSE phenotypes in both natural hosts and experimental animals.
In TSEs, there are no gross neuropathological lesions but there are characteristic histological lesions: bilateral and symmetrical widespread neuropil and/or neuronal vacuolation (spongiform appearance), synaptic changes, neuronal loss, gliosis, a variable degree and type of accumulation of PrPSc and sometimes amyloid plaques [1][2][3] (Figure 1). Spongiform changes are a general hallmark of TSEs; albeit neuronal vacuolation is frequent in animal prion diseases, it is uncommon in humans and has to be distinguished from non-specific spongiosis. This includes brain oedema, metabolic encephalopathies, autolysis and artefacts [1]. TSE spongiform degeneration is widespread or focal with small or oval uniform-sized, well-delineated and empty vacuoles in the neuropil and the neuron perikaryon. These changes can sometimes result from an artefact caused by the fixation and paraffin-embedded processing of the brain tissue [4]. It might also be caused by abnormal membrane permeability, autophagy or accumulation of PrPSc in the lysosomes [4]. Some studies revealed that vacuolation is not related to PrPSc accumulation [4].
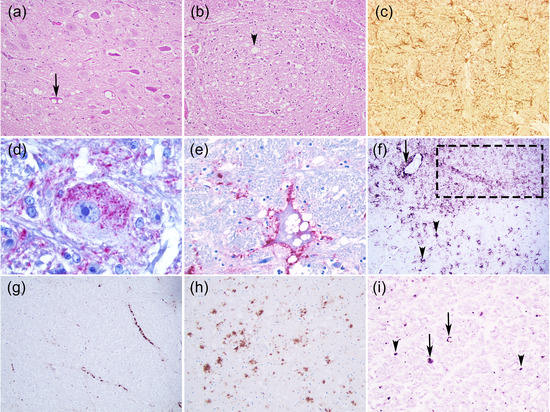
Figure 1. Neurohistopathological features of animal prion diseases. (a) Neuronal vacuolation (arrow) (Classical scrapie, sheep, medulla oblongata, dorsal vagal nucleus); (b) Neuropil vacuolation (arrow head) (Classical BSE, bovine, medulla oblongata, nucleus of the solitary tract); (c) Astrogliosis (Atypical scrapie, sheep, medulla oblongata, GFAP Polyclonal antibody, DAKO, 1:1000 dilution, x200); (d–i) Some PrPSc deposition types: (d) Intraneuronal (Feline spongiform encephalopathy, cat, medulla oblongata, reticular formation, 3F4 PrP Residues 109–111 Monoclonal antibody, DAKO, 1:300 dilution, x1000); (e) Perineuronal (BSE, bovine, medulla oblongata, reticular formation, 6H4 PrP Residues 144–152 Monoclonal antibody, Prionics AG, dilution 1:1000 x400); (f) Fine granular (dot line), stellate (arrow head), perivascular (arrow) (Classical scrapie, sheep, medulla oblongata, 2G11 PrP Residues 146-R154-R171-182 Monoclonal antibody, Pourquier Institute, 1:200 dilution, x100); (g) Linear (Classical BSE, bovine, medulla oblongata, reticular formation, 12F10 PrP Residues 142–160 Monoclonal antibody, SPBIO, 1:200 dilution, x200); (h) Plaque-like (cerebral cortex section from the CWD Proficiency testing 2008 organized by the European Reference Laboratory for TSEs-APHA, Weybridge, 2G11 PrP Monoclonal antibody, 1:200 dilution, x100; (i) Punctuate (arrow head) and globular (arrow) (Atypical scrapie, sheep, medula oblongata, pyramidal tract, 2G11 PrP Monoclonal antibody, 1:200 dilution). (a–b) Haematoxylin and eosin (H & E) stain, x200; (d–e) StreptABC-AP, Nova fucsina, Mayer’s Haematoxylin; (c, f–i) Vectastain Elite –HRP, DAB, Mayer’s Haematoxylin.
Gliosis and PrPSc deposition, not always associated with the severity of lesions, precede neuronal damage and neuropil vacuolation in the brain suggesting that a neuroinflammatory component might contribute to neuronal loss in these diseases [5]. Nevertheless, the mechanism of neuronal damage, the precise role of microgliosis and astrogliosis in that neurodegeneration and the cell tropism by a specific prion strain require additional research.
2. Prions and Animal Prion Diseases
Prion is defined as a “proteinaceous infectious particle” [6] consisting exclusively of a single protein without the involvement of nucleic acids that causes spongiform encephalopathies in mammals. Prion diseases are characterized by the accumulation of abnormal isoforms of PrP glycoprotein. Disease-associated isoform, designated PrPSc when protease-resistant, is derived from its physiological cellular prion protein precursor PrPC, by a posttranslational mechanism that involves conformational change and aggregation [7]. The pathogenic misfolded isoform is able to transmit misfolding by transforming more PrPC towards an increasing in β-sheet structure in detriment to the physiological α-helical structure and a tendency to aggregate into oligomers [8].
The prion protein gene (PRNP), highly conserved across mammals, is considered the major genetic determinant of susceptibility to prion diseases. Exon composition varies from one exon (e.g., Canis lupus familiaris, Felis catus, and Ovis aries), two exons (Homo sapiens), three exons (Cervus elaphus) or four exons (like Bos taurus), according to the Ensembl database. Nevertheless, a high level of conservation is observed in the coding sequence and aminoacidic sequence. A comparative genomic approach including nine species allows us to visualize the more conserved regions of the protein, as presented in Figure 2.

Figure 2. Multiple alignment of PrP protein from Ovis aries (ENSOARP00020014736), Capra hircus (CAA63050.1), Bos Taurus (ENSBTAP00000069134), Canis familiaris (ENSCAFP00000009107), Felis catus (ENSFCAP00000030786), Rangifer tarandus (ABS87897.1), Cervus elaphus (AAU93885.1), Alces alces (AZB50215.1) and Camelus dromedarius (CAA70901.1). The alignment was performed using T-Coffee program [9] and edited using Genedoc software [10].
Multiple mutations and polymorphisms of PRNP have been described in different animal species and some of them demonstrated an important effect on prion disease susceptibility/resistance. Figure 3 presents the missense mutations detected in PrP protein from Ovis aries. Variants are classified above and below the bar as ‘Risk factor’ and ‘Resistance’ according to in vivo and in vitro analyses [11][12][13][14]. The other variants were analysed by an in-silico approach through the PROVEAN algorithm (http://provean.jcvi.org/index.php, accessed on 27 January 2021) and classified as ”Deleterious” and ”Neutral”. All the missense mutations were gathered from Ensembl (ENSOART00020017839.1) and through database searching [13][14][15][16][17][18].
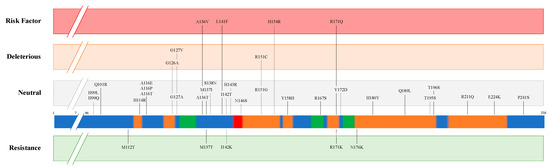
Figure 3. Missense mutations of prion protein (PrP) in Ovis aries. The coloured bar represents structural features of the protein according to UniProt (P23907), including α-helix (orange), β-strand (green) and turn (red). Variants are classified above and below the bar as ”Risk factor” and ”Resistance” according to in vivo and in vitro analyses. The other variants were analysed using an in-silico approach through PROVEAN algorithm, and classified as ”Deleterious” and ”Neutral”.
Different prion strains (isolates or variants) are distinct self-templating conformers [19] that display different phenotypes in a specified host and are preserved upon serial passage within the same host genotype. These distinct entities were first proposed in 1961 and since then have been characterized in different mammalian species with the implication in disease pathology and transmission [20]. Prion strains can be differentiated after experimental transmission to wild type or transgenic rodents by differences in the clinical signs, incubation period and the lesion profiles in the brain of the affected animals leading to the laboratory definition of a strain [3]. The lesion profile mainly focuses on differences in the regional patterns of prion-induced vacuolar neuropathology and/or PrPSc deposition but there are also strain differences in the association of PrPSc with particular brain cell types [21]. At the same time, biochemical characteristics of PrPSc as the electrophoretic mobility, protease resistance, glycosylation profile and sedimentation can be used to distinguish between prion strains [22].
As various distinct phenotypes can be identified in mice, the occurrence of different prion strains is known in classical scrapie (CS) of sheep and goats (revised by [23]), the first TSE described nearly 300 years ago, in a rare TSE in mink farms (TME) and chronic wasting disease (CWD) affecting farmed and free-ranging cervids, both identified in North America in 1947 and 1967, respectively (revised by [24]) (Figure 4). Contrarily, single strains have been associated with other animal prion diseases such as classical Bovine Spongiform Encephalopathy (C-BSE). C-BSE, responsible for an epidemic and a public health crisis in Europe, was identified in 1985 and largely increased the interest in the TSEs research due to its link to the variant Creutzfeldt-Jakob disease (vCJD) in humans and the putative transmission to other animal species (felids, zoo ruminants and small ruminants) [25]. This research and the reinforced TSE surveillance programs lead to the identification of more animal TSEs. As they showed distinct features from any TSE cases known at the time, the classical forms, they were designated as atypical (revised by [24]): atypical scrapie, atypical BSE and atypical CWD. Recently, a new prion disease was also confirmed in dromedaries—camel prion disease (CPD) (Figure 4). These could be new animal prion diseases or diseases not previously detected. Retrospective studies indicated that atypical scrapie has been present since at least 1972 [26].
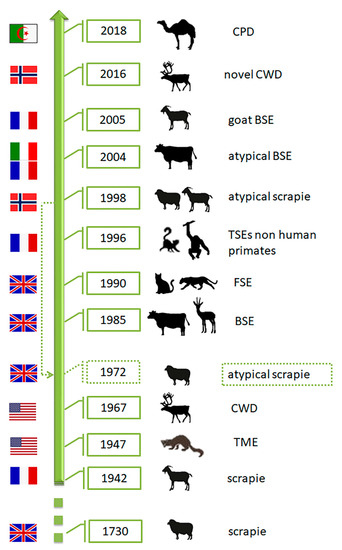
Figure 4. Chronology of identification of animal prion diseases. TME-transmissible mink encephalopathy; CWD—chronic wasting disease; BSE-bovine spongiform encephalopathy; FSE—feline spongiform encephalopathy; CPD—camel prion disease. The flags represent the country of the first report of the disease. The dashed arrow indicates retrospective identification of atypical scrapie in 1972 (Silhouettes from Freepik.com and img.inkfrog.com).
3. Neuroinflammation
Along with neurodegeneration, chronic neuroinflammation is a hallmark of prion brain diseases, but the process that leads to it is not yet fully understood. Recent research shows that during the accumulation of misfolded PrPSc both anti- and pro-inflammatory factors and molecules are active in the prion brain [27], reaching a stage of chronic inflammation that is likely to contribute to prion pathogenesis [5]. It appears that the vast majority of these factors is produced by cells within the CNS, namely microglial cells, cells that also exhibit PrPSc deposition [28], as it was demonstrated that the arrival of leucocytes from the periphery is limited and only detectable at the later stages of clinical disease [21][29][30]. As recorded for other diseases, microglia are the brain guardians, with several subpopulations, multifaceted, with neuroprotective but also neurotoxic properties [5], displaying region-dependent homeostatic transcriptional identities, observed during prion-associated neuroinflammation [31]. As such, the role of microglia in this group of diseases remains undefined, as they could be an actor in the pathogenesis or a simple consequence of the disease [32]. Besides, in scrapie, several chemokines were identified since the early asymptomatic stage and probably were enrolled in the disease progression. The inflammasome activation by the CD36 molecules also promotes the activation of several proinflammatory cytokines. All these factors (chemokines, inflammasome and proinflammatory cytokines) are crucial in the microglia recruitment and the inflammation and neuronal damage promoted by the PrPSc deposition [32]. Due to its nature and course, it is very difficult to address the neuroinflammation component of the disease in natural occurring prion cases. Thus, information regarding this aspect of the disease relies mainly on experimental studies that provide valuable and challenging data about the process.
During prion infection, and before reaching CNS, prions are first detected in lymphoid tissues, frequently associated with follicular dendritic cells (FDCs). They then progress through the nerves of the autonomic nervous system and finally reach the CNS where no apparent peripheral immune response is registered, albeit the activation of microglia and astrocytes [33]. In some experimental studies, the observation of increasing density of activated microglia cells, namely throughout the cerebellar cortex, as observed in natural cases, is associated with the upregulation of the TNF-α Receptor type-1 [34]. Single-cell transcriptional profiling screening 1/2 million cells revealed seven molecularly distinct and regionally restricted astrocyte types [35]. Astrocytes are the cells directly implicated in the direct neurotoxic effects related to the human and animal prions, namely the scrapie, and act as inflammation promoters [34]. In fact, astrocyte function pathway is activated in prion infection prior to the activated microglia or neuron and neurotransmission pathways [31].
Microgliosis is observed in infected brains even before the neuronal loss and spongiform change [5], microglial cells phagocytize prions and promote apoptotic cell clearance of neurons, a process mediated by the secretion of milk fat globule epidermal growth factor 8 (MFGE8) by astrocytes [36]. However, in vivo, this protective role of microglia becomes insufficient, which can induce microglia to convert from the phagocytic M2 phenotype into the pro-inflammatory M1 phenotype [37]. This altered phenotype may exacerbate the secretion of cytotoxic mediators and contributes to the spreading of prions, while increases the secretion of pro-inflammatory mediators by microglial cells [33].
Several studies allowed the identification of the inflammatory profile regarding the transcriptome and protein changes occurring in the prion- infected brain, some of them using scrapie strain infected mice [38]. Pro-inflammatory cytokines and chemokines, among which IL-1α and β, IL-12p40, TNF, CCL2–CCL6, and CXCL10, are increased in the brains of mice with the clinical disease [27][39]. High-density qRT-PC studies investigating different times of the disease progression revealed overexpression of inflammatory genes Il1rn [IL-1Ra], Ccl8 [CCL8/MCP-2], Tnfsf11 [Tnfsf11/RANKL], and Osm [OSM]) at the preclinical stage in scrapie strain 22L-infected mice). The upregulation of these genes occurred before clinical signs [40] and continued with disease progression. Later on, Oas1a, Isg15, Olr1, and Ccl5, among others, were found to be increased as well. Overall, the transcription of inflammatory mediators increases as the disease evolves to display the different clinical phases and endpoint [5]. The analysis of the transduction pathways in prion infected brain revealed that the AK-STAT and NF-κB pathways are substantially activated during the disease, being responsible for the transcription of many of these genes. Phosphorylated STAT proteins can act independently, but in addition they can also act synergistically with NF-κB [40], thus contributing to the enhancement of the expression of acute phase proteins such as haptoglobin, ceruloplasmin, α1-antichymotrypsin, and serum amyloid A [41][42].
On the other hand, several of the mentioned inflammatory mediators have the potential to induce damage to the CNS. The triggering of apoptosis in cells is controlled by the expression of Oas1a, Isg15, Tnfsf11, Olr1, and Ccl5 [43][44][45], cytokines like CCL2, CCL7, and CCL8 can attract monocytes [46], and it has also been shown that the expression of Cxcl10, Ccl2, A2m, and Tnf can contribute to neurotoxicity in other disease models [47][48][49][50].
The importance of region-specific astrocyte and microglial identities during prion- associated neuroinflammation was highlighted by recent studies: specific brain regions, as well as region-specific homeostatic identities are preserved during the preclinical stages of the disease. This is related to different prion strains and cell tropism. However, as disease progresses and the clinical signs arise, the region-specific homeostatic transcriptome signatures are replaced by the region-independent neuroinflammation signature, regardless of prion strain and cell tropism [31].These and other studies suggest that the inflammatory component of the disease concurs to neurodegeneration [39], by inducing cellular damage in the CNS and stimulating its surveillance system to display gliosis and activate astroglia and microglia observed in early disease before vacuolar pathology or clinical signs [21]. Ultimately, an inflammatory self-perpetuation cycle is then established in the prion infected brain, in which neuronal damage and astrocyte and microglial activation concur to the pathobiology of the disease and its outcome. The increase microgliosis is closely associated with spongiosis and astrogliosis, and the correct understanding of its functions must be, in the future, related with therapies, including the microglia reprogram throughout the prion clearance mechanisms, instead of neurotoxicity associated with these cell populations in the later stages of prion diseases [5].
References
- Budka, H. Neuropathology of Prion Diseases. Br. Med. Bull. 2003, 66, 121–130.
- VanDeVelde, M.; Higgins, R.J.; Oevermann, A. Veterinary Neuropathology: Essentials of Theory and Practice; Wiley-Blackwell: Oxford, UK, 2012; ISBN 978-0-470-67056-9.
- Spiropoulos, J.; Simmons, M.M. Pathology of Animal Transmissible Spongiform Encephalopathies (TSEs). Food Saf. 2017, 5, 1–9.
- Soto, C.; Satani, N. The Intricate Mechanisms of Neurodegeneration in Prion Diseases. Trends Mol. Med. 2011, 17, 14–24.
- Carroll, J.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65.
- Prusiner, S. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144.
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833.
- Jucker, M.; Walker, L.C. Self-Propagation of Pathogenic Protein Aggregates in Neurodegenerative Diseases. Nature 2013, 501, 45–51.
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment 1. J. Mol. Biol. 2000, 302, 205–217.
- Nicholas, K.B.; Nicholas, H.B.J. Deerfield D.W.GeneDoc Analysis and Visualization of Genetic Variation. EMBnet News 1997, 4, 1–4.
- Vaccari, G.; D’Agostino, C.; Nonno, R.; Rosone, F.; Conte, M.; Di Bari, M.A.; Chiappini, B.; Esposito, E.; De Grossi, L.; Giordani, F.; et al. Prion Protein Alleles Showing a Protective Effect on the Susceptibility of Sheep to Scrapie and Bovine Spongiform Encephalopathy. J. Virol. JVI 2007, 81, 7306–7309.
- Laegreid, W.W.; Clawson, M.L.; Heaton, M.P.; Green, B.T.; O’Rourke, K.I.; Knowles, D.P. Scrapie Resistance in ARQ Sheep. J. Virol. 2008, 82, 10318–10320.
- Greenlee, J.J. Review: Update on Classical and Atypical Scrapie in Sheep and Goats. Vet. Pathol. 2019, 56, 6–16.
- Cassmann, E.D.; Moore, S.J.; Smith, J.D.; Greenlee, J.J. Sheep With the Homozygous Lysine-171 Prion Protein Genotype Are Resistant to Classical Scrapie After Experimental Oronasal Inoculation. Vet. Pathol. 2019, 56, 409–417.
- DeSilva, U.; Guo, X.; Kupfer, D.M.; Fernando, S.C.; Pillai, A.T.V.; Najar, F.Z.; So, S.; Fitch, G.Q.; Roe, B.A. Allelic Variants of Ovine Prion Protein Gene (PRNP) in Oklahoma Sheep. Cytogenet. Genome Res. 2003, 102, 89–94.
- Kdidi, S.; Yahyaoui, M.H.; Conte, M.; Chiappini, B.; Zaccaria, G.; Ben Sassi, M.; Ben Ammar El Gaaied, A.; Khorchani, T.; Vaccari, G. PRNP Polymorphisms in Tunisian Sheep Breeds. Livest. Sci. 2014, 167, 100–103.
- Kim, S.-K.; Kim, Y.-C.; Won, S.-Y.; Jeong, B.-H. Potential Scrapie-Associated Polymorphisms of the Prion Protein Gene (PRNP) in Korean Native Black Goats. Sci. Rep. 2019, 9, 15293.
- Teferedegn, E.Y.; Yaman, Y.; Ün, C. Novel Variations in Native Ethiopian Goat Breeds PRNP Gene and Their Potential Effect on Prion Protein Stability. Sci. Rep. 2020, 10, 6953.
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational Variations in an Infectious Protein Determine Prion Strain Differences. Nature 2004, 428, 323–328.
- Morales, R.; Abid, K.; Soto, C. The Prion Strain Phenomenon: Molecular Basis and Unprecedented Features. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 681–691.
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551.
- Morales, R. Prion Strains in Mammals: Different Conformations Leading to Disease. PLoS Pathog. 2017, 13.
- Nonno, R.; Marin-Moreno, A.; Carlos Espinosa, J.; Fast, C.; Van Keulen, L.; Spiropoulos, J.; Lantier, I.; Andreoletti, O.; Pirisinu, L.; Di Bari, M.A.; et al. Characterization of Goat Prions Demonstrates Geographical Variation of Scrapie Strains in Europe and Reveals the Composite Nature of Prion Strains. Sci. Rep. 2020, 10, 19.
- Aguilar-Calvo, P.; García, C.; Espinosa, J.C.; Andreoletti, O.; Torres, J.M. Prion and Prion-like Diseases in Animals. Virus Res. 2015, 207, 82–93.
- Sigurdson, C.J.; Miller, M.W. Other Animal Prion Diseases. Br. Med. Bull. 2003, 66, 199–212.
- Chong, A.; Kennedy, I.; Goldmann, W.; Green, A.; González, L.; Jeffrey, M.; Hunter, N. Archival Search for Historical Atypical Scrapie in Sheep Reveals Evidence for Mixed Infections. J. Gen. Virol. 2015, 96, 3165–3178.
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J. Virol. 2016, 90, 3003–3017.
- Andréoletti, O.; Lacroux, C.; Chabert, A.; Monnereau, L.; Tabouret, G.; Lantier, F.; Berthon, P.; Eychenne, F.; Lafond-Benestad, S.; Elsen, J.-M.; et al. PrPSc Accumulation in Placentas of Ewes Exposed to Natural Scrapie: Influence of Foetal PrP Genotype and Effect on Ewe-to-Lamb Transmission. J. Gen. Virol. 2002, 83, 2607–2616.
- Baker, C.A.; Manuelidis, L. Unique Inflammatory RNA Profiles of Microglia in Creutzfeldt-Jakob Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679.
- Lewicki, H.; Tishon, A.; Homann, D.; Mazarguil, H.; Laval, F.; Asensio, V.C.; Campbell, I.L.; DeArmond, S.; Coon, B.; Teng, C.; et al. T Cells Infiltrate the Brain in Murine and Human Transmissible Spongiform Encephalopathies. J. Virol. 2003, 77, 3799–3808.
- Makarava, N.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. Region-Specific Glial Homeostatic Signature in Prion Diseases Is Replaced by a Uniform Neuroinflammation Signature, Common for Brain Regions and Prion Strains with Different Cell Tropism. Neurobiol. Dis. 2020, 137, 104783.
- Ano, Y.; Sakudo, A.; Onodera, T. Effect of Microglial Inflammation in Prion Disease. Curr. Issues Mol. Biol. 2020, 1–12.
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The Immunobiology of Prion Diseases. Nat. Rev. Immunol. 2013, 13, 888–902.
- Ragagnin, A.; Ezpeleta, J.; Guillemain, A.; Boudet-Devaud, F.; Haeberlé, A.-M.; Demais, V.; Vidal, C.; Demuth, S.; Béringue, V.; Kellermann, O.; et al. Cerebellar Compartmentation of Prion Pathogenesis: Prions in Cerebellum. Brain Pathol. 2018, 28, 240–263.
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22.
- Kranich, J.; Krautler, N.J.; Falsig, J.; Ballmer, B.; Li, S.; Hutter, G.; Schwarz, P.; Moos, R.; Julius, C.; Miele, G.; et al. Engulfment of Cerebral Apoptotic Bodies Controls the Course of Prion Disease in a Mouse Strain–Dependent Manner. J. Exp. Med. 2010, 207, 2271–2281.
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the Degenerating Brain Are Capable of Phagocytosis of Beads and of Apoptotic Cells, but Do Not Efficiently Remove PrPSc, Even upon LPS Stimulation. Glia 2010, 58, 2017–2030.
- Xiang, W.; Windl, O.; Wünsch, G.; Dugas, M.; Kohlmann, A.; Dierkes, N.; Westner, I.M.; Kretzschmar, H.A. Identification of Differentially Expressed Genes in Scrapie-Infected Mouse Brains by Using Global Gene Expression Technology. J. Virol. 2004, 78, 11051–11060.
- Crespo, I.; Roomp, K.; Jurkowski, W.; Kitano, H.; del Sol, A. Gene Regulatory Network Analysis Supports Inflammation as a Key Neurodegeneration Process in Prion Disease. BMC Syst. Biol. 2012, 6, 132.
- Carroll, J.A.; Striebel, J.F.; Race, B.; Phillips, K.; Chesebro, B. Prion Infection of Mouse Brain Reveals Multiple New Upregulated Genes Involved in Neuroinflammation or Signal Transduction. J. Virol. 2015, 89, 2388–2404.
- Uskokovic, A.; Dinic, S.; Mihailovic, M.; Grigorov, I.; Ivanovic-Matic, S.; Bogojevic, D.; Grdovic, N.; Arambasic, J.; Vidakovic, M.; Martinovic, V.; et al. STAT3/NFκB Interplay in the Regulation of Α2-Macroglobulin Gene Expression During Rat Liver Development and the Acute Phase Response. IUBMB Life 2017, 59, 170–178.
- Bode, J.G.; Albrecht, U.; Häussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic Acute Phase Proteins—Regulation by IL-6- and IL-1-Type Cytokines Involving STAT3 and Its Crosstalk with NF-ΚB-Dependent Signaling. Eur. J. Cell Biol. 2012, 91, 496–505.
- Castelli, J.C.; Hassel, B.A.; Maran, A.; Paranjape, J.; Hewitt, J.A.; Li, X.; Hsu, Y.-T.; Silverman, R.H.; Youle, R.J. The Role of 2′-5′ Oligoadenylate-Activated Ribonuclease L in Apoptosis. Cell Death Differ. 1998, 5, 313–320.
- Valerio, A.; Ferrario, M.; Martinez, F.O.; Locati, M.; Ghisi, V.; Bresciani, L.G.; Mantovani, A.; Spano, P. Gene Expression Profile Activated by the Chemokine CCL5/RANTES in Human Neuronal Cells. J. Neurosci. Res. 2004, 78, 371–382.
- Potu, H.; Sgorbissa, A.; Brancolini, C. Identification of USP18 as an Important Regulator of the Susceptibility to IFN-α and Drug-Induced Apoptosis. Cancer Res. 2010, 70, 655–665.
- Van Coillie, E.; Van Damme, J.; Opdenakker, G. The MCP/Eotaxin Subfamily of CC Chemokines. Cytokine Growth Factor Rev. 1999, 10, 61–86.
- Fabrizi, C.; Businaro, R.; Lauro, G.M.; Starace, G.; Fumagalli, L. Activated Α2Macroglobulin Increases β-Amyloid (25–35)-Induced Toxicity in LAN5 Human Neuroblastoma Cells. Exp. Neurol. 1999, 155, 252–259.
- Kovacs, D. Α2-Macroglobulin in Late-Onset Alzheimer’s Disease. Exp. Gerontol. 2000, 35, 473–479.
- Sui, Y.; Stehno-Bittel, L.; Li, S.; Loganathan, R.; Dhillon, N.K.; Pinson, D.; Nath, A.; Kolson, D.; Narayan, O.; Buch, S. CXCL10-Induced Cell Death in Neurons: Role of Calcium Dysregulation. Eur. J. Neurosci. 2006, 23, 957–964.
- Severini, C.; Passeri, P.P.; Ciotti, M.; Florenzano, F.; Possenti, R.; Zona, C.; Di Matteo, A.; Guglielmotti, A.; Calissano, P.; Pachter, J.; et al. Bindarit, Inhibitor of CCL2 Synthesis, Protects Neurons Against Amyloid-β-Induced Toxicity. J. Alzheimers Dis. 2013, 38, 281–293.


