 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Riccardo Rizzo | + 2640 word(s) | 2640 | 2021-03-17 05:13:23 | | | |
| 2 | Peter Tang | Meta information modification | 2640 | 2021-03-20 06:03:16 | | | | |
| 3 | Donatella Delle Cave | Meta information modification | 2640 | 2021-03-22 14:04:48 | | | | |
| 4 | Donatella Delle Cave | Meta information modification | 2640 | 2021-03-22 17:29:48 | | | | |
| 5 | Donatella Delle Cave | Meta information modification | 2640 | 2021-03-22 17:35:13 | | |
Video Upload Options
Pancreatic cancer is an extremely lethal malignancy with a survival rate lower than any other cancer type. For decades, two-dimensional (2D) cultures have been the cornerstone for studying cancer cell biology and drug testing, due to their simplicity and cost. However, their inability to reconstitute the tumor architecture, the absence of nutrient and oxygen supply gradients, as well as the lack of appropriate mechano-forces that mimic the extracellular microenvironment, make them an inadequate model to accurately reproduce tissue level-specific characteristics. Bioengineering systems, such as three-dimensional (3D) patient-specific models, are progressively emerging as systems better able to mimic the biology of pancreatic tumors and to test new anticancer therapies, as they more efficiently recapitulate the complex tumor microenvironment characteristic of pancreatic tumors.
1. Introduction
Pancreatic cancer (PC) is a devastating and essentially incurable disease, leading to patient death in the majority of cases [1]. Incidence and mortality are increasing steadily, and PC is predicted to become the second leading cause of cancer-related death by 2030 [2]. According to the Italian Association of Medical Oncology (AIOM), in 2019, approximately 13,500 new PC cases were diagnosed in Italy [3]. These dramatic numbers are due largely to late diagnosis, lack of effective therapies, and a poor understanding of PC biology. Thus, PC has become a world healthcare priority, and studies focused on novel in vitro models to investigate tumor progression and develop more effective treatments are urgently needed. One reason for the lack of success for the majority of drugs used to treat PC patients is their inappropriate purposing and associated toxicity. At the preclinical stage, two-dimensional (2D) cultures have been a milestone in the study of cancer biology, since they represent a useful platform for analyzing genetic and molecular alterations and a cost-effective system for drug screening [4]. However, they are an inherently and extremely simplified model that fail to precisely reflect the human tumor microenvironment (TME) and its molecular components [5], which can inevitably lead to non-translatable results [6][7]. Those aspects are crucial for the study of PC, characterized by an abundant desmoplastic core, which accounts for up to 90% of the total tumor volume, and by an intricate cross-talk among tumor and stromal cell, both of which are critical aspects for cancer progression [8][9]. The TME is a very complex ecosystem in which several cellular (such as pancreatic stellate cells (PSCs), cancer-associated fibroblasts (CAFs), and immune cells) and non-cellular components (such as cytokines, immunoregulatory molecules, and extracellular matrix (ECM) components) are involved, contributing to the development of a hypoxic and “cold” immunosuppressive tumor, resistant to chemotherapy, targeted therapy, and immunotherapy [10]. While the number of drugs targeting the TME is progressively increasing, there is an urgent demand for more biologically and physiologically relevant in vitro preclinical models that can accurately mimic the TME that exists in patient tumors. The limitations of 2D models have been, in part, overcome by the use of genetically engineered mouse models (GEMMs), which recreate the most frequent genetic alterations associated with pancreatic cancer progression and provide a more physiological microenvironment for drug testing and genetic research [11]. However, in vivo studies are expensive, time-consuming, and ethically not suitable for drug screening and, most important, the results obtained have limited relevance in humans [12]. The opportunity of reproducing the TME in vitro by growing tumor cells in three-dimensional (3D) matrixes or scaffolds has opened new horizons in the field of drug testing. These 3D in vitro tumor models have been shown to be superior to 2D models, allowing for multiple cell populations to interact and mimicking the complexity and the biomechanical properties of tumors as well as tumor heterogeneity, and allowing for treatment responses similar to those seen in patients diagnosed with PC [13][14][15][16][17].
2. 3D In Vitro Models
3D in vitro systems are advantageous predictive tools, which may accelerate translating basic research into personalized medicine by providing more physiological information on cellular responses to different stimuli [18].
2.1. Spheroid
Spheroids are the simplest 3D model, which can be generated by the hanging drop technique, consisting of embedding cells in different matrices (such as collagen, methylcellulose, gelatin, and alginate hydrogels), or by growing single-cell suspensions in ultra-low attachment plates, in order to minimize the adhesion to plastic supports and to optimize cell–cell and cell–matrix interactions [19]. Spheroids can arise from self-assembling cells or via cellular aggregation and constitute an easy and highly reproducible 3D model [20]. Recently, Cavo and colleagues developed a new method to generate spheroids from the pancreatic ductal adenocarcinoma (PDAC) cell line (i.e., MiaPaCa2) with the hanging drop technique. They used a methylcellulose-enriched media and hydrophobic substrates to generate well-organized pancreatic spheroids with increased mesenchymal and cancer stem cell features [21]. This method appears to be efficient for the long-term generation of spheroids, even from those cell lines that hardly give rise to 3D structures due to low cohesiveness (Figure 1a). By comparing 2D cultures with 3D spheroids generated by different techniques (e.g., ultra-low adhesion concave microwells, Matrigel inclusion, and organotypic systems), Zeeberg and colleagues found differences in cell growth, morphology, and in the response to different stimuli. They concluded that the organotypic culture, generated by plating cells on the top of a matrix gel, more closely recapitulated the tissue architecture of PC [22].
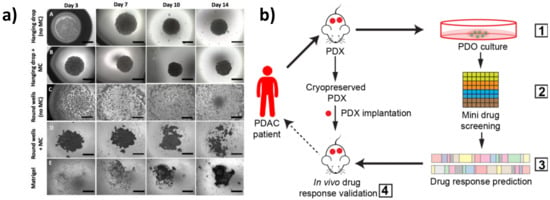
Figure 1. Three-dimensional (3D) in vitro models of pancreatic cancer. (a) MiaPaCa-2 spheroid growth using different methods. Scale bars = 1000 μm (adapted from [21]). (b) Schematic representation of the patient-derived organoid (PDO)-derived system. (1) Generation of PDO and patient-derived xenograft (PDX) models from cryopreserved xenografts of patients with pancreatic ductal adenocarcinoma (PDAC). Screening of Food and Drug Administration (FDA)-approved drugs (2) and validation in organoid cultures (3). Selection of effective drugs for small-scale drug screenings and validation in the PDXs (4) (adapted from [23]).
2.2. Patient-Derived Organoids (PDOs)
PDOs are 3D structures established from freshly isolated primary cells, which retain the ability of self-renewal and self-organization in an organ-like structure that recapitulates the tissue of origin. A pioneer study in establishing pancreatic organoids from both normal and freshly isolated human tumor biopsies was performed by Boj et al. [24]. The authors optimized the standard culture condition protocols for generating organoids able to recapitulate disease-specific alterations, enabling them to recreate in vitro the different stages of tumor progression. Specifically, single-cell suspensions are embedded in a Matrigel or collagen matrix and supplemented with a media containing a well-defined combination of tissue-specific growth factors [24][25][26]. PDOs can be genetically engineered in vitro by CRISPR-Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats) technology to specifically edit tumor-driving genes, and can be xenografted in immunodeficient mice for in vivo studies [27].
The potential of pancreatic PDOs has also been recently exploited for the development of a PDO-based gene expression signature [28] and for the establishment of PDO biobanks [29]. PDOs represent a powerful model to recapitulate tumor histology and for personalized drug screening. Intriguingly, Seino and colleagues identified three functional PDO subtypes in PDAC on the basis of differences in stem cell niche factor dependency. By engineering them with clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 technology, they found that exogenous WNT is required to support the initial stages of tumorigenesis but it is dispensable in the late stage of tumor progression [30]. Pancreatic PDOs have also been shown to be as effective at determining drug efficacy compared to more labor-intensive and cost-prohibitive in vivo patient-derived xenograft (PDX) models. For example, Frappart et al. published a small-scale drug screening PDO platform that was successfully validated in an in vivo xenograft trial, highlighting the clinical utility of PDOs for validating and discovering new treatments for PC [23] (Figure 1b). Moreover, Nelson et al. used different multidisciplinary approaches, which included confocal microscopy and transcription profiling analysis, to compare PDX organoids with isogenetically matched 2D primary cell lines from PDAC tumors and primary cell line organoids (CLOs) [31]. Interestingly, they developed an in vitro method to generate CLOs, which very closely recapitulated the main features of organoids generated from PDXs, thus overcoming the limitations associated with xenografts. Along these lines, PDO drug screening platforms have recently been improved by the development of automated organoid-based platforms that take advantage of microfluidic systems for the simultaneous and time-controlled test of thousands of compounds, enabling real-time genetic and phenotypic analysis of PDOs, serving as a step forward in personalized therapy [32].
3. Reconstitution of Tumor Cell Heterogeneity and Complexity in 3D In Vitro Models
PC is characterized by a dense desmoplastic reaction defined by different cellular components, such as immune cells, fibroblasts, endothelial cells, and PSCs, dispersed in an organized extracellular matrix enriched in collagens, hyaluronic acid, and laminins [33][34]. The TME is therefore conditioned by distinct environmental factors, i.e., cytokines, growth factors, as well as a specific biochemical profile, which mediate cellular communication and are crucial in influencing cancer progression [35][36]. Reconstituting the tumor complexity by using 3D models requires taking into consideration the specific interactions between matrix components and the different cellular types present in the tumor in order to recreate in vitro the specific environmental conditions that distinguish each tumor. Attempts to recreate such a complex environment is also one of the ways to master in vitro tumor modeling.
4. Investigating Cell–Cell Interactions in In Vitro 3D Models
4.1. 3D Bioprinting
The use of scaffolds to generate 3D models has opened the way for innovative techniques such as 3D bioprinting. Cell printing combines scaffolds and different cell types to create a complex model with precise structure and high reproducibility [37]. Bioprinting facilitates a controlled spatial and temporal distribution of cells [38]. A 3D bioprinted tumor model can be generated by different techniques: inkjet printing, extrusion-based printing (EBP), laser-assisted bioprinting (LAB), and stereolithography [39]. 3D bioprinting has recently been used by Langer et al. to form multicellular structures consisting first of a PDAC cell core surrounded by primary human PSCs and umbilical vein endothelial cells (HUVECs). The authors demonstrated that multi-cell-type bioprinted tissues can recapitulate aspects of the in vivo tumor and provide a tunable system for the examination of several tumorigenic endpoints in the distinct tumor microenvironments [39]. Hakobyan et al. established a spheroid-based array using 3D bioprinting technology capable of reproducing the different stages of PDAC development in order to improve the understanding of PC tumor biology. This model allowed them to induce in vitro acinar to ductal cell transdifferentiation, a crucial process in PDAC progression [40].
Nevertheless, one of the major limitations of these 3D models is the lack of vasculature, which not only provides a supply of oxygen and nutrients but is essential for cancer metastasis. To overcome this limitation, microfluidics has emerged as a cutting-edge technology for combining the cellular reproducibility of PDOs with the flow control of a tumor-on-a-chip platform [41].
4.2. Organ-on-A-Chip
Although it is clear that tumors are heterogeneous mixtures of cells and ECM components, the extent to which different cell types influence cell–cell interactions as well as the paracrine signaling that is generated during therapy remains poorly understood. Nowadays, numerous microscopy-based imaging techniques are available to analyze cell morphology and cell–cell interactions within in vitro tumor models, including confocal microscopy, two-photon microscopy, and light sheet fluorescence microscopy [42][43][44]. One of the main advantages of using microscopy-based imaging approaches is the ability to observe spatial relations among different cell types with high temporal resolution under physiological conditions [45]. In this scenario, in vitro PDAC-on-a-chip provides powerful platforms to study the microenvironment of PDAC since these devices allow imaging of cell–cell interactions, such as tumor–endothelial and tumor–immune cell interactions, as well as cell morphological changes, by applying different types of fluorescence microscopy techniques [4][46][47][48][49][50]. In a recent work by Hye-ran Moon et al., a microfluidic pancreatic tumor model was developed, recapitulating the heterogeneous driver mutations of human PCs by using PDAC cells derived from genetically engineered mouse models (KPC with Kras and Trp53 mutations, and KIC with Kras mutation and Cdkn2a deletion) in order to mimic the intra-tumoral heterogeneity (Figure 2a) [49]. The model was successfully used to study interaction mechanisms between heterogeneous cancer cell subpopulations exposed to anti-cancer drugs and associated drug resistance. By means of fluorescence microscopy, the authors observed significant morphological changes in the epithelial phenotype KIC (eKIC) cells co-cultured with the mesenchymal phenotype KIC (mKIC) cells at the level of enhanced gemcitabine resistance in the co-culture models (Figure 2b), suggesting that interactions between these two cancer cell types induced multiple changes of the eKIC cells including loss of epithelial characteristics, most likely causing increased resistance to gemcitabine. By means of confocal immunofluorescence, the authors assessed the changes in E-cadherin (E-cad) expression and observed that when co-culturing cells, E-cad expression in eKIC was significantly reduced, implying that the interactions between heterogeneous cancer cells may induce the phenotype transition of epithelial cancer cell types. In a different approach, Nguyen and colleagues reported a new organotypic PDAC-on-a-chip model that mimics vascular invasion and tumor–blood vessel interactions (Figure 2c–f) [51]. The microfluidic device is composed of two hollow cylindrical channels embedded within a 3D collagen matrix. One channel is seeded with endothelial cells to form a perfusable biomimetic blood vessel, while the other channel is seeded with PC cells to form a pancreatic cancer duct. To study the interactions of PDAC cells with the blood vessels, the authors performed a screening experiment wherein different chemotactic agents were introduced into the biomimetic blood vessel and found that a gradient of fetal bovine serum most efficiently stimulated the invasion of PC cells into the collagen matrix. By means of confocal microscopy, the authors were able to record the ablation of blood vessel by cancer cells—notably, they observed that once in contact with the biomimetic blood vessel, the PDAC cells wrapped around the blood vessel and spread along the length of the blood vessel before invading into the vessel itself, leaving behind tumor-lined and tumor-filled luminal structures (Figure 2c-f). The infiltration of immunosuppressive cells is critical in the generation and maintenance of an immunosuppressive environment in PDAC, thus contributing to the failure of current available therapeutic approaches. In this regard, cell–cell interaction studies have revealed an important interplay between tumor-associated macrophages and regulatory T cells (Tregs). By using light sheet fluorescent microscopy, Siret C. et al. were able to show a direct interaction between myeloid-derived suppressor cells (MDSCs) and Tregs cells in a 3D PDAC tumor context [52]. These findings were also corroborated by the use of the transwell system, which demonstrated that cell-to-cell interactions are required for Treg cell proliferation and development induced by MDSCs. However, Treg cells were also shown to modulate proliferation and survival of MDSCs. Remarkably, by coupling imaging approaches and functional assays, the authors were able to show that physical interactions between cells contribute to the establishment of an immunosuppressive environment in PDAC.
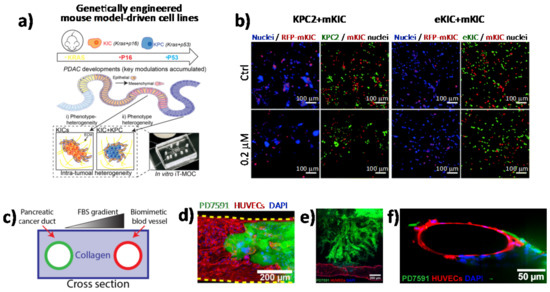
Figure 2. Direct imaging of cell–cell interactions in 3D in vitro models of PC. (a) Schematic illustration of the functional model of the in vitro intra-tumoral heterogeneous features composed by genetically engineered mouse model-driven cell lines to capture different PDAC progression stages. (b) Representative fluorescent micrographs of co-cultured KPC2–mKIC and eKIC–mKIC in control (Ctrl), 0.2 μM, and 20 μM gemcitabine treatment groups. Nuclei (blue) of each cell are distinguished in green (KPC2 and eKIC) and red (mKIC). Abbreviations: iT-MOC (interstitial tumor-microenvironment-on-a-chip), KIC genotypes (Kras mutation and Cdkn2a deletion), mKIC (mesenchymal phenotype KIC), murine pancreatic cancer cell lines (KPC2, eKIC, and mKIC). (a,b) (adapted from [49]). (c) Schematic illustration of PDAC-on-a-chip with a biomimetic blood vessel and a PC duct. One channel is seeded with endothelial cells to form a perfusable biomimetic blood vessel, while the other channel is seeded with PC cells to form a tumor duct. FBS (fetal bovine serum). (d) Representative confocal image of a section of the blood vessel (in red) invaded by YFP PD7591 PDAC cell (in green), showing that part of the blood vessel is being ablated by cancer cells in the organotypic model. (e) YFP PD7591 cells (in green) invading the biomimetic blood vessel (in red), migrating along the vessel and wrapping around the blood vessel. (f) Cross-sectional image of the biomimetic blood vessel shown in (e). (c-e) (adapted from [51]).
References
- Fitzmaurice, C. Global Burden of Disease Cancer Collaboration. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 2006 to 2016: A systematic analysis for the Global Burden of Disease study. J. Clin. Oncol. 2018, 36, 1568.
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921.
- Silvestris, N.; Brunetti, O.; Bittoni, A.; Cataldo, I.; Corsi, D.; Crippa, S.; D’Onofrio, M.; Fiore, M.; Giommoni, E.; Milella, M.; et al. Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up of Exocrine Pancreatic Ductal Adenocarcinoma: Evidence Evaluation and Recommendations by the Italian Association of Medical Oncology (AIOM). Cancers 2020, 12, 1681.
- Swayden, M.; Soubeyran, P.; Iovanna, J. Upcoming Revolutionary Paths in Preclinical Modeling of Pancreatic Adenocarcinoma. Front. Oncol. 2020, 9, 1443.
- Michalski, C.W.; Rosendahl, J.; Michl, P.; Kleeff, J. Translational Pancreatic Cancer Research: From Understanding of Mechanisms to Novel Clinical Trials. In Molecular and Translational Medicine; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-030-49475-9.
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-Derived Xenografts Undergo Murine-Specific Tumor Evolution. Nat. Genet. 2017, 49, 1567.
- Barros, A.S.; Costa, E.C.; Nunes, A.S.; De Melo-Diogo, D.; Correia, I.J. Comparative study of the therapeutic effect of Doxorubicin and Resveratrol combination on 2D and 3D (spheroids) cell culture models. Int. J. Pharm. 2018, 551, 76–83.
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 1–15.
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325.
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Westphalen, C.B.; Olive, K.P. Genetically Engineered Mouse Models of Pancreatic Cancer. Cancer J. 2012, 18, 502–510.
- Gopinathan, A.; Morton, J.P.; Jodrell, D.I.; Sansom, O.J. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis. Model. Mech. 2015, 8, 1185–1200.
- Tomás-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074–5089.
- Ehlen, L.; Arndt, J.; Treue, D.; Bischoff, P.; Loch, F.N.; Hahn, E.M.; Kotsch, K.; Klauschen, F.; Beyer, K.; Margonis, G.A.; et al. Novel methods for in vitro modeling of pancreatic cancer reveal important aspects for successful primary cell culture. BMC Cancer 2020, 20, 1–13.
- Cavo, M.; Serio, F.; Kale, N.R.; D’Amone, E.; Gigli, G.; Del Mercato, L.L. Electrospun nanofibers in cancer research: From engineering of in vitro 3D cancer models to therapy. Biomater. Sci. 2020, 8, 4887–4905.
- Polini, A.; Del Mercato, L.L.; Barra, A.; Zhang, Y.S.; Calabi, F.; Gigli, G. Towards the development of human immune-system-on-a-chip platforms. Drug Discov. Today 2019, 24, 517–525.
- Turetta, M.; Del Ben, F.; Brisotto, G.; Biscontin, E.; Bulfoni, M.; Cesselli, D.; Colombatti, A.; Scoles, G.; Gigli, G.; Del Mercato, L.L. Emerging Technologies for Cancer Research: Towards Personalized Medicine with Microfluidic Platforms and 3D Tumor Models. Curr. Med. Chem. 2018, 25, 4616–4637.
- Kimlin, L.; Kassis, J.; Virador, V. 3Din vitrotissue models and their potential for drug screening. Expert Opin. Drug Discov. 2013, 8, 1455–1466.
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95.
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6.
- Cavo, M.; Cave, D.D.; D’Amone, E.; Gigli, G.; Lonardo, E.; Del Mercato, L.L. A synergic approach to enhance long-term culture and manipulation of MiaPaCa-2 pancreatic cancer spheroids. Sci. Rep. 2020, 10, 1–11.
- Zeeberg, K.; Cardone, R.A.; Greco, M.R.; Saccomano, M.; Nøhr-Nielsen, A.; Alves, F.; Pedersen, S.F.; Reshkin, S.J. Assessment of different 3D culture systems to study tumor phenotype and chemosensitivity in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2016, 49, 243–252.
- Frappart, P.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids—A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 8, 594–606.
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338.
- Hwang, C.-I.; Boj, S.F.; Clevers, H.; Tuveson, D.A. Preclinical models of pancreatic ductal adenocarcinoma. J. Pathol. 2016, 238, 197–204.
- Moreira, L.; Bakir, B.; Chatterji, P.; Dantes, Z.; Reichert, M.; Rustgi, A.K. Pancreas 3D Organoids: Current and Future Aspects as a Research Platform for Personalized Medicine in Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 289–298.
- Fujii, M.; Clevers, H.; Sato, T. Modeling Human Digestive Diseases with CRISPR-Cas9–Modified Organoids. Gastroenterology 2019, 156, 562–576.
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129.
- Driehuis, E.; Van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590.
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.
- Nelson, S.R.; Zhang, C.; Roche, S.; O’Neill, F.; Swan, N.; Luo, Y.; Larkin, A.; Crown, J.; Walsh, N. Modelling of pancreatic cancer biology: Transcriptomic signature for 3D PDX-derived organoids and primary cell line organoid development. Sci. Rep. 2020, 10, 1–12.
- Schuster, B.; Junkin, M.; Kashaf, S.S.; Romero-Calvo, I.; Kirby, K.; Matthews, J.; Weber, C.R.; Rzhetsky, A.; White, K.P.; Tay, S. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 2020, 11, 1–12.
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540.
- Cave, D.D.; Di Guida, M.; Costa, V.; Sevillano, M.; Ferrante, L.; Heeschen, C.; Corona, M.; Cucciardi, A.; Lonardo, E. TGF-β1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene 2020, 39, 4271–4285.
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276.
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676.
- Mandrycky, C.; Wang, Z.; Kim, K.; Kim, D.-H. 3D bioprinting for engineering complex tissues. Biotechnol. Adv. 2016, 34, 422–434.
- Asghar, W.; El Assal, R.; Shafiee, H.; Pitteri, S.; Paulmurugan, R.; Demirci, U. Engineering cancer microenvironments for in vitro 3-D tumor models. Mater. Today 2015, 18, 539–553.
- Langer, E.M.; Allen-Petersen, B.L.; King, S.M.; Kendsersky, N.D.; Turnidge, M.A.; Kuziel, G.M.; Riggers, R.; Samatham, R.; Amery, T.S.; Jacques, S.L.; et al. Modeling Tumor Phenotypes In Vitro with Three-Dimensional Bioprinting. Cell Rep. 2019, 26, 608–623.
- Hakobyan, D.; Médina, C.; Dusserre, N.; Stachowicz, M.-L.; Handschin, C.; Fricain, J.-C.; Guillermet-Guibert, J.; Oliveira, H. Laser-assisted 3D bioprinting of exocrine pancreas spheroid models for cancer initiation study. Biofabrication 2020, 12, 035001.
- Lai, B.F.L.; Lu, R.X.Z.; Hu, Y.; Huyer, L.D.; Dou, W.; Wang, E.Y.; Radulovich, N.; Tsao, M.S.; Sun, Y.; Radisic, M. Recapitulating Pancreatic Tumor Microenvironment through Synergistic Use of Patient Organoids and Organ-on-a-Chip Vasculature. Adv. Funct. Mater. 2020.
- Conchello, J.-A.; Lichtman, J.W. Optical sectioning microscopy. Nat. Methods 2005, 2, 920–931.
- Mertz, J. Optical sectioning microscopy with planar or structured illumination. Nat. Methods 2011, 8, 811–819.
- Reynaud, E.G.; Kržič, U.; Greger, K.; Stelzer, E.H.K. Light sheet-based fluorescence microscopy: More dimensions, more photons, and less photodamage. HFSP J. 2008, 2, 266–275.
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845.
- Beer, M.; Kuppalu, N.; Stefanini, M.; Becker, H.; Schulz, I.; Manoli, S.; Schuette, J.; Schmees, C.; Casazza, A.; Stelzle, M.; et al. A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: Comparison with in vitro cultures and in vivo xenografts. Sci. Rep. 2017, 7, 1–12.
- Bradney, M.J.; Venis, S.M.; Yang, Y.; Konieczny, S.F.; Han, B. A Biomimetic Tumor Model of Heterogeneous Invasion in Pancreatic Ductal Adenocarcinoma. Small 2020, 16, e1905500.
- Bi, Y.; Shirure, V.S.; Liu, R.; Cunningham, C.; Ding, L.; Meacham, J.M.; Goedegebuure, S.P.; George, S.C.; Fields, R.C. Tumor-on-a-chip platform to interrogate the role of macrophages in tumor progression. Integr. Biol. 2020, 12.
- Moon, H.-R.; Ozcelikkale, A.; Yang, Y.; Elzey, B.D.; Konieczny, S.F.; Han, B. An engineered pancreatic cancer model with intra-tumoral heterogeneity of driver mutations. Lab Chip 2020, 20, 3720–3732.
- Kramer, B.; De Haan, L.; Vermeer, M.; Olivier, T.; Hankemeier, T.; Vulto, P.; Joore, J.; Lanz, H.L. Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins. Int. J. Mol. Sci. 2019, 20, 4647.
- Nguyen, D.-H.T.; Lee, E.; Alimperti, S.; Norgard, R.J.; Wong, A.; Lee, J.J.-K.; Eyckmans, J.; Stanger, B.Z.; Chen, C.S. A biomimetic pancreatic cancer on-chip reveals endothelial ablation via ALK7 signaling. Sci. Adv. 2019, 5, eaav6789.
- Siret, C.; Collignon, A.; Silvy, F.; Robert, S.; Cheyrol, T.; André, P.; Rigot, V.; Iovanna, J.; Van De Pavert, S.; Lombardo, D.; et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2020, 10, 3070.



