 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yeri Rim | + 2328 word(s) | 2328 | 2020-04-03 05:02:37 | | | |
| 2 | Nicole Yin | -5 word(s) | 2323 | 2020-10-27 10:45:26 | | |
Video Upload Options
Osteoarthritis (OA) is the most common joint disease that causes pain and disability in the adult population. OA is primarily caused by trauma induced by an external force or by age-related cartilage damage. Chondrocyte hypertrophy or chondrocyte senescence is thought to play a role in the initiation and progression of OA. Although chondrocyte hypertrophy and cell death are both crucial steps during the natural process of endochondral bone formation, the abnormal activation of these two processes after injury or during aging seems to accelerate the progression of OA. However, the exact mechanisms of OA progression and these two processes remain poorly understood. Chondrocyte senescence and hypertrophy during OA share various markers and processes. In this study, we reviewed the changes that occur during chondrocyte hypertrophy or senescence in OA and the attempts that were made to regulate them. Regulation of hypertrophic or senescent chondrocytes might be a potential therapeutic target to slow down or stop OA progression; thus, a better understanding of the processes is required for management.
1. Introduction
Chondrocyte hypertrophy and cell death are natural phenomena that usually occur during a developmental process called EO. Hypertrophic chondrocytes appear and play a crucial role in EO. Hyaline cartilage can be divided into two groups: 1) temporary and 2) permanent cartilage. Healthy cartilage is usually called permanent cartilage or resting chondrocytes, which are present in the articulating joint. Usually, permanent cartilage has a low proliferation rate and does not undergo terminal differentiation and EO under normal conditions[1]. Temporary cartilage is initially formed as cartilage, but the final product is bone. Unrestricted differentiation of precursor cells into the chondrocyte lineage does not lead to permanent cartilage, but bone[1]. Chondrocytes undergo active proliferation and generate a cascade of cells, whereas some of them undergo enlargement, and others hypertrophical changes and become hypertrophic chondrocytes. These cells increase their volume dramatically, and the surroundings become mineralized to develop bone tissue[2]. The elastic nature of cartilage begins to change and harden through calcification. This makes it more difficult for the chondrocytes to receive nutrients, as most of the cells undergo apoptosis and leave small cavities within the tissue, which leaves enough room in the hardened bone for blood vessel invasion. Through this process, the cartilage turns into trabecular bone. However, the major highlight events of EO, such as chondrocyte proliferation, hypertrophic differentiation of chondrocytes, cell death, calcification or mineralization, blood vessel invasion, and chondrocyte apoptosis, equivalently occur in OA (Figure 1).
2. Hypertrophy and Senescence-Related Markers
Cell hypertrophy generally refers to an increase in cell size and volume. Hypertrophic differentiation of chondrocytes can also be characterized by the high expression of collagen type X, runt-related transcription factor 2 (RUNX2) and MMP13. Hyaline cartilage markers, such as aggrecan, collagen type II, and SOX9, are decreased in the hypertrophic cells. Although hypertrophical changes in chondrocytes are required in bone growth and development, the mechanism acts as a double-edged sword in diseased environments[3].
Hypertrophic differentiation is also an important pathological change in OA cartilage[4]. Cartilage tissue fractures at sites that receive high mechanical stress, which is a process called fibrillation[5]. During fibrillation in the cartilage, the tissue loses several ECM proteins, including proteoglycans and collagen fibers. After these changes, the subchondral bone generates osteophytes. Abnormal chondrocyte hypertrophy in articular cartilage is seen in cartilage destruction, which is a potential therapeutic target for OA treatment[6][7]. Hypertrophic chondrocytes start to express genes related to osteogenic differentiation, and start to produce mineralized ECM proteins[8][9]. It is unclear if the abnormally differentiated hypertrophic chondrocytes give rise to fully differentiated bone cells, however, apoptosis of hypertrophic chondrocytes were suggested in previous studies. Lacunar emptying within OA cartilage was observed, which leaded to the loss of the articular cartilage[10][11]. After these changes, the subchondral bone generates osteophytes. Chondrocyte calcification by calcium deposition is the final stage of chondrocyte hypertrophy[1]. Previous studies reported that the layer bordering the calcified cartilage and the non-calcified cartilage increases after the age of 60 years[12]. Although this layer of calcified cartilage tends to increase throughout the aging process, in some cases, the calcified region decreases instead. This indicated that the lower calcified cartilage was replaced by bone due to the EO-like process in OA.
Several markers are known as chondrocyte hypertrophy and senescence markers. Both hypertrophy and senescence are not usually characterized by a specific set of markers, but are rather associated with a set of cellular phenotypes that often co-exist in a stressed environment (Figure 1)[13]. Although both chondrocyte hypertrophy and senescence are not clearly understood, markers such as IL-1β, collagen type X, RUNX2, vascular endothelial growth factor (VEGF), osteopontin, osteocalcin, and Indian hedgehog (IHH) are thought to be related[14][15]. The accumulation of MMPs induced by pro-inflammatory cytokines is also a positive senescence marker in OA chondrocytes.
Collagen type X is a major marker used to detect chondrocyte hypertrophy. This type of collagen is usually not expressed in healthy articular cartilage, though it is thought to play a role in the early stage of endochondral bone formation since it has been detected at sites of hypertrophic chondrocyte regions and calcification. Protein and mRNA levels of collagen type X are also detected in human OA cartilage but their expression shows significant local variation[1][6][16][17][18]. Despite the overall expression of collagen type X, alpha 1 (COL10A1) was increased in human OA cartilage, and the expression was higher in the relatively less degenerated area[19]. Isolated chondrocytes cultured in vitro lose their normal morphology and start synthesizing collagen type X after several passages[20][21]. For example, whereas passage 2 chondrocytes show high levels of collagen type II and low levels of collagen type X, passage 6 chondrocytes show the opposite characteristics with increased collagen type X[14]. The exact mechanism of senescence and collagen type X is not fully understood, but its expression is thought to be the main reason for chondrocyte dedifferentiation[22].
Elevated levels of MMP13 have been reported in OA cartilage[23][24]. The gene level of MMP13 was highly expressed in OA cartilage when compared to normal levels. Elevated expression of MMP13 and collagen type X was reported after inducing knee joint stability by the transection of knee ligaments and meniscus removal in mouse OA models[25]. MMP13 is also thought to play a central role in the irreversible degradation of collagen type II in OA[26][27]. Knockdown of activin-like kinase 5 (ALK5) was reported to increase MMP13 mRNA levels[28]. TGFβ is important for the maintenance and protection of healthy articular cartilage; therefore, it is commonly used in the in vitro culture of chondrocytes or chondrogenesis. The binding of TGFβ to its receptor recruits the type I receptor ALK5. In turn, this molecule complex phosphorylates the smad2 and 3 in the smad2/3 pathway, which is known to repress chondrocyte terminal differentiation[29][30]. Another alternative receptor ALK1 activates the smad1/5/8 signaling pathway, which is known to induce chondrocyte terminal differentiation[31][32]. The change in the ALK1/ALK5 ratio during aging or OA development may result in increased dominance of the smad1/5/8 signaling in chondrocytes and induce a hypertrophic-like state by high levels of MMP13. MMP13 is also a central regulator of chondrocyte senescence[33]. Chondrocytes undergoing senescence produce ECM-degrading enzymes such as MMP13, which promotes the degradation of the main protein of the articular cartilage, aggrecan, and collagen type II. MMP13 produced by cultured chondrocytes isolated from older donors appeared to be increased[34]. Changes in chondrocyte function (e.g., decreased anabolic activity and increased catabolic activity) are thought to be related to MMP13 production.
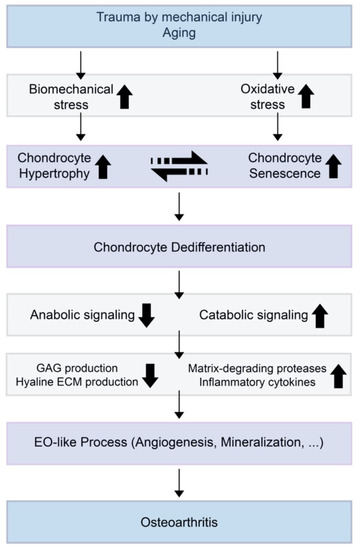
Figure 1. A theoretical model for the relationships of chondrocyte hypertrophy and chondrocyte senescence and the initiation or progression of osteoarthritis (OA). When trauma is induced by mechanical injury or aging, biomechanical stress and oxidative stress increase in the cartilage environment. Increased chondrocyte hypertrophy and senescence induce decreased anabolic signaling and increased catabolic signaling. This results in suppressed glycosaminoglycan (GAG) production by normal chondrocytes and increases the expression of extracellular matrix (ECM)-degrading enzymes and inflammatory cytokines.
RUNX2 is the main transcription factor that is involved in hypertrophic chondrocyte differentiation and early osteogenesis[24][35]. One of the hallmarks of OA is the upregulation of RUNX2. Therefore, RUNX2 is assumed to be a major transcriptional factor that directly regulates the expression of matrix degradation enzymes in the damaged articular cartilage[36]. When the destabilization of the medial meniscus (DMM) osteoarthritis model was induced in RUNX2 knockout mice, the gene expression of matrix degradation enzymes (i.e., MMP9, MMP13, ADAMTS4, ADAMTS5, ADAMTS7, and ADAMTS12) was significantly reduced compared with DMM-induced Cre-negative control. The deletion of RUNX2 in DMM-induced mice decreased MMP13 protein levels in the articular cartilage. Cells expressing ectopic RUNX2 showed a senescent-like phenotype that was characterized by an enlarged and flattened morphology and β-galactosidase staining; p53 signaling was required for this process[37].
A characteristic feature of hypertrophy and OA cartilage is the increased production of VEGF. VEGF induces the migration of endothelial cells by chemotactic actions and induces angiogenesis in vivo. VEGF also promotes angiogenesis in the cartilage tissue, which is related to the calcification of chondrocytes that can lead to dysregulated osteogenesis of the normal cartilage. Neoangiogenesis in the cartilage growth plate plays an important role in EO; therefore, VEGF is thought of as a critical mediator during EO. Carlevaro et al. investigated the expression of VEGF in mammalian and avian embryo long bone growth plates[38]. Although VEGF was observed in fully mature hypertrophic chondrocytes, it was completely absent in proliferating and quiescent cells in both chicken and mice. VEGF mRNA generates five different isoforms with a different number of amino acid residues by alternative splicing, labeled VEGF121, VEGF145, VEGF165, VEGF189, and VEGF206[39]. Although only three types (VEGF121, VEGF165, and VEGF189) were detectable in both OA and normal cartilage, their receptors were only found in OA cartilage. OA cartilage secreted a significantly higher amount of VEGF than normal cartilage when cultured in vitro. VEGF expression is induced by mechanical forces on articular cartilage[40]. Therefore, it is thought that VEGF may regulate chondrocyte differentiation in response to mechanical forces[41]. After inducing DMM in mice, immunostaining showed increased levels of VEGF in articular cartilage, subchondral bone, synovium, and meniscus.
IHH is also suggested as a marker for chondrocyte hypertrophy. IHH is a key signaling molecule that is expressed in pre-hypertrophic chondrocytes during growth plate development and that regulates chondrocyte hypertrophy during EO[42]. The upregulation of IHH in postnatal cartilage showed an increased chondrocyte hypertrophy and degradation[43]. Activation of IHH downstream signaling pathways results in a reduction in articular cartilage thickness and proteoglycan content levels, while inhibition of IHH signaling reversed these effects[44][45]. IHH is reported to regulate the onset of chondrocyte hypertrophy by a negative feedback loop with parathyroid hormone-related protein (PTHrP)[46]. IHH activates the expression of PTHrP in particular chondrocytes and PTHrP signals through the receptor PTHR1 to inhibit chondrocyte hypertrophy and maintain the proliferating state. Mak et al. reported that IHH signaling can also directly regulate chondrocyte hypertrophy in the absence of PTHrP. IHH also promoted chondrocyte hypertrophy through Wnt/β-catenin activation and bone morphogenetic proteins (BMPs) signaling.
Expressions of p16, p21, and p53 increase in senescent cells, which result in cell cycle arrest. Articular chondrocytes with increased expression of p16 and p21 exhibit increased senescence. Chondrocytes expressing p53 showed similar morphology to OA chondrocytes and underwent apoptosis, whereas downregulation of p53 expression prevented chondrocytes from undergoing senescence or apoptosis[47]. Chondrocytes expressing p16 lose their normal phenotype and express increased inflammatory cytokines and MMPs such as IL-1β, IL-6, and MMP13[22]. In chondrocytes isolated from cadaveric donors without evidence of OA between 17 to 72 years old, age was found to be responsible for 27% of the variation in p16 levels. However, loss of p16 was not critical enough to prevent the development of age-induced or injury-induced traumatic OA[48]. Collagen type II expression is negatively correlated with p21 expression[49].
Oxidative stress caused by ROS is a major regulator in cell senescence. ROS induce multiple genes that spark senescence or dedifferentiation in chondrocytes. The main ROS reported to induce cartilage damage in chondrocytes are hydrogen peroxide (H2O2) and peroxynitrite[50][51]. Degenerated cartilage regions in OA patients had significantly lower anti-oxidative potential than the intact cartilage region, confirming increased oxidative damage in degenerated cartilage[52]. Chondrocytes treated with H2O2 showed shorter telomere length in vitro. When OA cartilage tissue was cultured with H2O2 addition, the number of glycosaminoglycans (GAGs) gradually decreased in a time-dependent manner. However, the addition of an anti-oxidative agent reversed these effects, inhibited GAG loss, and maintained telomere length. H2O2 addition significantly increased p21 expression in both early and late passage chondrocytes[53]. The expression started to increase one hour after treatment and reached its highest level after three hours, and the change in early passage chondrocytes was nine-fold higher than the initial level.
Growth factors such as BMPs and other cytokines are reported to play a role in chondrocyte hypertrophy. Several BMPs enhance chondrogenesis; however, dysregulated levels of BMPs may induce calcification of cartilage and accelerate cartilage degradation. During OA development, chondrocytes reportedly express BMPs [54][55]. The BMP canonical smad1/5/8 pathway is thought of as an inducer of chondrocyte hypertrophy and EO[56]. Cytokines such as IL-6 and IL-8 are known to induce growth arrest of senescent cells when secreted into the cell microenvironment. IL-1β is associated with chondrocyte dedifferentiation, and induces repression of SOX9 and the synthesis of nitric oxide (NO)[14][57]. The induction of NO by IL-1β activates c-Jun N-terminal kinases (JNK) signaling, which is a well-known inducer of senescence[58]. IL-7 is a cytokine that is upregulated age-dependently[59]. The expression of IL-7 is higher in cultured articular chondrocytes from older donors compared to that of the younger donors. Furthermore, increased levels of MMP13 are induced in chondrocytes after IL-7 treatment[60].
Advanced glycation end products (AGEs) are produced from reducing sugars and free amino groups of proteins, lipids, or nucleic acids by a non-enzymatic reaction[61][62]. AGE accumulation suppresses collagen type II production and also stimulates the expression of MMPs and ADAMTSs[63][64][65]. Collagen cross-linking is increased by AGEs, which enhances tissue stiffness[66][67]. Increased AGEs are pathogenic and induce oxidative stress and inflammation. AGEs also induce the secretion of TNF-α, and NO[64][68] .
References
- P.M. Van Der Kraan; W.B. Van Den Berg; Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration?. Osteoarthritis and Cartilage 2012, 20, 223-232, 10.1016/j.joca.2011.12.003.
- E.J. Mackie; Y.A. Ahmed; L. Tatarczuch; K.-S. Chen; M. Mirams; Endochondral ossification: How cartilage is converted into bone in the developing skeleton. The International Journal of Biochemistry & Cell Biology 2008, 40, 46-62, 10.1016/j.biocel.2007.06.009.
- C. C. Van Donkelaar; W. Wilson; Mechanics of chondrocyte hypertrophy. Biomechanics and Modeling in Mechanobiology 2011, 11, 655-664, 10.1007/s10237-011-0340-0.
- Chengjie Lian; Xudong Wang; Xianjian Qiu; Zizhao Wu; Bo Gao; Lei Liu; Guoyan Liang; Hang Zhou; XiaoMing Yang; Yan Peng; et al.Anjing LiangCaixia XuDongsheng HuangPeiqiang Su Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin β1−SMAD1 interaction. Bone Research 2019, 7, 1-15, 10.1038/s41413-019-0046-y.
- Ok Hee Jeon; Nathaniel David; Judith Campisi; Jennifer H. Elisseeff; Senescent cells and osteoarthritis: a painful connection. Journal of Clinical Investigation 2018, 128, 1229-1237, 10.1172/jci95147.
- Kirsch, T.; Swoboda, B.; Nah, H. Activation of annexin II and V expression, terminal differentiation, mineralization and apoptosis in human osteoarthritic cartilage. Osteoarthr. Cartil. 2000, 8, 294–302.
- Tchetina, E.V.; Kobayashi, M.; Yasuda, T.; Meijers, T.; Pidoux, I.; Poole, A.R. Chondrocyte hypertrophy can be induced by a cryptic sequence of type II collagen and is accompanied by the induction of MMP-13 and collagenase activity: Implications for development and arthritis. Matrix Biol. 2007, 26, 247–258.
- Findlay, D.M.; Atkins, G.J. Osteoblast-chondrocyte interactions in osteoarthritis. Curr. Osteoporos Rep. 2014, 12, 127–134.
- Li, J.; Dong, S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016, 2016, 2470351
- Sharif, M.; Whitehouse, A.; Sharman, P.; Perry, M.; Adams, M. Increased apoptosis in human osteoarthritic cartilage corresponds to reduced cell density and expression of caspase-3. Arthritis Rheum. 2004, 50, 507–515.
- Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collee, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146.
- L B Lane; P G Bullough; Age-related changes in the thickness of the calcified zone and the number of tidemarks in adult human articular cartilage.. null 1980, 62, 372–375.
- Kendal McCulloch; Gary J. Litherland; Taranjit Singh Rai; Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210-218, 10.1111/acel.12562.
- S Salman Ashraf; Byung Hyo Cha; J.-S. Kim; J. Ahn; Ilwoo Han; H. Park; S.-H. Lee; Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis and Cartilage 2016, 24, 196-205, 10.1016/j.joca.2015.07.008.
- Kristen A. Johnson; Deborah Van Etten; Nisha Nanda; Robert M. Graham; Robert A. Terkeltaub; Distinct Transglutaminase 2-independent and Transglutaminase 2-dependent Pathways Mediate Articular Chondrocyte Hypertrophy. Journal of Biological Chemistry 2003, 278, 18824-18832, 10.1074/jbc.m301055200.
- Hennig, T.; Lorenz, H.; Thiel, A.; Goetzke, K.; Dickhut, A.; Geiger, F.; Richter, W. Reduced chondrogenic potential of adipose tissue derived stromal cells correlates with an altered TGFbeta receptor and BMP profile and is overcome by BMP-6. J. Cell Physiol. 2007, 211, 682–691.
- Pullig, O.; Weseloh, G.; Ronneberger, D.; Kakonen, S.; Swoboda, B. Chondrocyte differentiation in human osteoarthritis: Expression of osteocalcin in normal and osteoarthritic cartilage and bone. Calcif. Tissue Int. 2000, 67, 230–240.
- Aigner, T.; Fundel, K.; Saas, J.; Gebhard, P.M.; Haag, J.; Weiss, T.; Zien, A.; Obermayr, F.; Zimmer, R.; Bartnik, E. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006, 54, 3533–3544.
- Naoshi Fukui; Yasuko Ikeda; Toshiyuki Ohnuki; Nobuho Tanaka; Atsuhiko Hikita; Hiroyuki Mitomi; Toshihito Mori; Takuo Juji; Yozo Katsuragawa; Seizo Yamamoto; et al.Motoji SawabeShoji YamaneRyuji SuzukiLinda J. SandellTakahiro Ochi Regional differences in chondrocyte metabolism in osteoarthritis: A detailed analysis by laser capture microdissection. Arthritis & Rheumatism 2007, 58, 154-163, 10.1002/art.23175.
- David G. Stokes; Gang Liu; Rita Dharmavaram; David Hawkins; Sonsoles Piera-Velazquez; Sergio A. Jimenez; Regulation of type-II collagen gene expression during human chondrocyte de-differentiation and recovery of chondrocyte-specific phenotype in culture involves Sry-type high-mobility-group box (SOX) transcription factors. Biochemical Journal 2001, 360, 461-470, 10.1042/bj3600461.
- Byung-Hyun Cha; Ji-Seon Lee; Sung Won Kim; Hyuk-Jin Cha; Soo-Hong Lee; The modulation of the oxidative stress response in chondrocytes by Wip1 and its effect on senescence and dedifferentiation during in vitro expansion. Biomaterials 2013, 34, 2380-2388, 10.1016/j.biomaterials.2012.12.009.
- Didier Philipot; David Guérit; Daniela Platano; Paul Chuchana; Eleonora Olivotto; Francisco Espinoza; Anne Dorandeu; Yves-Marie Pers; Jacques Piette; Rosa Maria Borzi'; et al.Christian JorgensenDanièle NoëlJean-Marc Brondello p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Research & Therapy 2014, 16, R58-R58, 10.1186/ar4494.
- Shlopov, B.V.; Gumanovskaya, M.L.; Hasty, K.A. Autocrine regulation of collagenase 3 (matrix metalloproteinase 13) during osteoarthritis. Arthritis Rheum. 2000, 43, 195–205.
- Wang, X.; Manner, P.A.; Horner, A.; Shum, L.; Tuan, R.S.; Nuckolls, G.H. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 963–973.
- S. Kamekura; K. Hoshi; T. Shimoaka; U. Chung; H. Chikuda; T. Yamada; M. Uchida; N. Ogata; A. Seichi; K. Nakamura; et al.H. Kawaguchi Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis and Cartilage 2005, 13, 632-641, 10.1016/j.joca.2005.03.004.
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 2001, 107, 35–44.
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733.
- Esmeralda N. Blaney Davidson; Dennis F. G. Remst; Elly L. Vitters; Henk M. Van Beuningen; Arjen B. Blom; Marie-Jose Goumans; Wim B. Van Den Berg; Peter M. Van Der Kraan; Increase in ALK1/ALK5 Ratio as a Cause for Elevated MMP-13 Expression in Osteoarthritis in Humans and Mice. The Journal of Immunology 2009, 182, 7937-7945, 10.4049/jimmunol.0803991.
- Ferguson, C.M.; Schwarz, E.M.; Reynolds, P.R.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. Smad2 and 3 mediate transforming growth factor-beta1-induced inhibition of chondrocyte maturation. Endocrinology 2000, 141, 4728–4735.
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46.
- Goumans, M.J.; Mummery, C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol. 2000, 44, 253–265.
- Ito, H.; Akiyama, H.; Shigeno, C.; Nakamura, T. Noggin and bone morphogenetic protein-4 coordinately regulate the progression of chondrogenic differentiation in mouse clonal EC cells, ATDC5. Biochem. Biophys. Res. Commun. 1999, 260, 240–244.
- Wei Zhang; Chi Zhang; Congfeng Luo; Yulin Zhan; Biao Zhong; Design, cyclization, and optimization of MMP13–TIMP1 interaction-derived self-inhibitory peptides against chondrocyte senescence in osteoarthritis. International Journal of Biological Macromolecules 2019, 121, 921-929, 10.1016/j.ijbiomac.2018.10.141.
- Christopher B. Forsyth; Ada Cole; Gillian Murphy; Julia L. Bienias; Hee-Jeong Im; Richard F. Loeser; Increased Matrix Metalloproteinase-13 Production With Aging by Human Articular Chondrocytes in Response to Catabolic Stimuli. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2005, 60, 1118-1124, 10.1093/gerona/60.9.1118.
- J. Hecht; V. Seitz; M. Urban; F. Wagner; P.N. Robinson; A. Stiege; C. Dieterich; U. Kornak; U. Wilkening; N. Brieske; et al.C. ZwingmanA. KidessS. StrickerS. Mundlos Detection of novel skeletogenesis target genes by comprehensive analysis of a Runx2−/− mouse model. Gene Expression Patterns 2007, 7, 102-112, 10.1016/j.modgep.2006.05.014.
- Di Chen; Jie Shen; Weiwei Zhao; Tingyu Wang; Lin Han; John L Hamilton; Hee-Jeong Im; Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Research 2017, 5, 16044, 10.1038/boneres.2016.44.
- Anna Kilbey; Karen Blyth; Sandy Wotton; Anne Terry; Alma Jenkins; Margaret Bell; Linda Hanlon; Ewan R. Cameron; James C. Neil; Runx2 disruption promotes immortalization and confers resistance to oncogene-induced senescence in primary murine fibroblasts.. Cancer Research 2007, 67, 11263-11271, 10.1158/0008-5472.CAN-07-3016.
- M F Carlevaro; S Cermelli; R Cancedda; Fiorella Descalzi Cancedda; Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: auto-paracrine role during endochondral bone formation.. Journal of Cell Science 2000, 113, 59–69.
- Hiroyuki Enomoto; Isao Inoki; Koichiro Komiya; Takayuki Shiomi; Eiji Ikeda; Ken-Ichi Obata; Hideo Matsumoto; Yoshiaki Toyama; Yasunori Okada; Vascular Endothelial Growth Factor Isoforms and Their Receptors Are Expressed in Human Osteoarthritic Cartilage. The American Journal of Pathology 2003, 162, 171-181, 10.1016/s0002-9440(10)63808-4.
- Rainer Beckmann; Astrid Houben; M. Tohidnezhad; N. Kweider; Athanassios Fragoulis; Christoph Jan Wruck; Lars-Ove Brandenburg; Benita Hermanns-Sachweh; Mary B Goldring; Thomas Pufe; et al.Holger Jahr Mechanical Forces Induce Changes in VEGF and VEGFR-1/sFlt-1 Expression in Human Chondrocytes. International Journal of Molecular Sciences 2014, 15, 15456-15474, 10.3390/ijms150915456.
- Masashi Nagao; John L. Hamilton; Ranjan Kc; Agnes D. Berendsen; Xuchen Duan; Chan Wook Cheong; Xin Li; Hee-Jeong Im; Bjorn R. Olsen; Vascular Endothelial Growth Factor in Cartilage Development and Osteoarthritis. Scientific Reports 2017, 7, 1-16, 10.1038/s41598-017-13417-w.
- Fangyuan Wei; Jingming Zhou; Xiaochun Wei; Juntao Zhang; Braden C. Fleming; Richard Terek; Ming Pei; Qian Chen; Tao Liu; Lei Wei; et al. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthritis and Cartilage 2012, 20, 755-763, 10.1016/j.joca.2012.03.010.
- Alvin C Lin; Brian L Seeto; Justyna M Bartoszko; Michael A Khoury; Heather Whetstone; Louisa Ho; Claire Hsu; S Amanda Ali; Benjamin A Alman; Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nature Medicine 2009, 15, 1421-1425, 10.1038/nm.2055.
- Beaupre, G.S.; Stevens, S.S.; Carter, D.R. Mechanobiology in the development, maintenance, and degeneration of articular cartilage. J. Rehabil. Res. Dev. 2000, 37, 145–151.
- Mak, K.K.; Kronenberg, H.M.; Chuang, P.T.; Mackem, S.; Yang, Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008, 135, 1947–1956.
- Benoit St-Jacques; Matthias Hammerschmidt; Andrew P. McMahon; Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes & Development 1999, 13, 2072-2086, 10.1101/gad.13.16.2072.
- Shingo Hashimoto; Takayuki Nishiyama; Shinya Hayashi; Takaaki Fujishiro; Ken Takebe; Noriyuki Kanzaki; Ryosuke Kuroda; Masahiro Kurosaka; Role of p53 in human chondrocyte apoptosis in response to shear strain. Arthritis & Rheumatism 2009, 60, 2340-2349, 10.1002/art.24706.
- Brian O. Diekman; Garrett A. Sessions; John A. Collins; Anne K. Knecht; Susan L. Strum; Natalia K. Mitin; Cathy S. Carlson; Richard F. Loeser; Norman E. Sharpless; Expression of p16INK 4a is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 2018, 17, e12771, 10.1111/acel.12771.
- Hyeon Joo Kim; So Ra Park; Heon Joo Park; Byung Hyune Choi; Byoung-Hyun Min; Potential Predictive Markers for Proliferative Capacity of Cultured Human Articular Chondrocytes: PCNA and p21. Artificial Organs 2005, 29, 393-398, 10.1111/j.1525-1594.2005.29066.x.
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026.
- Minguzzi, M.; Cetrullo, S.; D’Adamo, S.; Silvestri, Y.; Flamigni, F.; Borzi, R.M. Emerging Players at the Intersection of Chondrocyte Loss of Maturational Arrest, Oxidative Stress, Senescence and Low-Grade Inflammation in Osteoarthritis. Oxid. Med. Cell Longev. 2018, 2018, 3075293.
- Kazuo Yudoh; Nguyen Van Trieu; Hiroshi Nakamura; Kayo Masuko; Tomohiro Kato; Kusuki Nishioka; Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Research & Therapy 2005, 7, R380-R391, 10.1186/ar1499.
- Anita Brandl; Andreas Hartmann; Volker Bechmann; Bernhard Graf; Michael Nerlich; Peter Angele; Oxidative stress induces senescence in chondrocytes. Journal of Orthopaedic Research 2011, 29, 1114-1120, 10.1002/jor.21348.
- Hemanth Akkiraju; Anja Nohe; Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. Journal of Developmental Biology 2015, 3, 177-192, 10.3390/jdb3040177.
- T. Nakase; T. Miyaji; T. Tomita; M. Kaneko; K. Kuriyama; A. Myoui; K. Sugamoto; T. Ochi; H. Yoshikawa; Localization of bone morphogenetic protein-2 in human osteoarthritic cartilage and osteophyte. Osteoarthritis and Cartilage 2003, 11, 278-284, 10.1016/s1063-4584(03)00004-9.
- Kelsey N. Retting; Buer Song; Byeong S. Yoon; Karen M. Lyons; BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093-1104, 10.1242/dev.029926.
- Amy L. McNulty; Bradley T. Estes; Rebecca E. Wilusz; J. Brice Weinberg; Farshid Guilak; Dynamic loading enhances integrative meniscal repair in the presence of interleukin-1. Osteoarthritis and Cartilage 2010, 18, 830-838, 10.1016/j.joca.2010.02.009.
- R. Clancy; J. Rediske; C. Koehne; D. Stoyanovsky; A. Amin; M. Attur; K. Iyama; S.B. Abramson; Activation of stress-activated protein kinase in osteoarthritic cartilage: evidence for nitric oxide dependence. Osteoarthritis and Cartilage 2001, 9, 294-299, 10.1053/joca.2000.0388.
- Meredith A. Greene; Richard F. Loeser; Aging-related inflammation in osteoarthritis. Osteoarthritis and Cartilage 2015, 23, 1966-1971, 10.1016/j.joca.2015.01.008.
- David Long; Simon Blake; Xiao-Yu Song; Michael Lark; Richard F. Loeser; Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Research & Therapy 2008, 10, R23-R23, 10.1186/ar2376.
- Zhang, W.; Randell, E.W.; Sun, G.; Likhodii, S.; Liu, M.; Furey, A.; Zhai, G. Hyperglycemia-related advanced glycation end-products is associated with the altered phosphatidylcholine metabolism in osteoarthritis patients with diabetes. PLoS ONE 2017, 12, e0184105.
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916 e912.
- Huang, C.Y.; Lai, K.Y.; Hung, L.F.; Wu, W.L.; Liu, F.C.; Ho, L.J. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology (Oxford) 2011, 50, 1379–1389.
- Nah, S.S.; Choi, I.Y.; Lee, C.K.; Oh, J.S.; Kim, Y.G.; Moon, H.B.; Yoo, B. Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology (Oxford) 2008, 47, 425–431.
- Nah, S.S.; Choi, I.Y.; Yoo, B.; Kim, Y.G.; Moon, H.B.; Lee, C.K. Advanced glycation end products increases matrix metalloproteinase-1, -3, and -13, and TNF-alpha in human osteoarthritic chondrocytes. FEBS Lett. 2007, 581, 1928–1932.
- Chen, A.C.; Temple, M.M.; Ng, D.M.; Verzijl, N.; DeGroot, J.; TeKoppele, J.M.; Sah, R.L. Induction of advanced glycation end products and alterations of the tensile properties of articular cartilage. Arthritis Rheum. 2002, 46, 3212–3217.
- Bank, R.A.; Bayliss, M.T.; Lafeber, F.P.; Maroudas, A.; Tekoppele, J.M. Ageing and zonal variation in post-translational modification of collagen in normal human articular cartilage. The age-related increase in non-enzymatic glycation affects biomechanical properties of cartilage. Biochem. J. 1998, 330 (Pt 1), 345–351.
- Feng-Cheng Liu; Li-Feng Hung; Wan-Lin Wu; Deh-Ming Chang; Chuan-Yueh Huang; Jenn-Haung Lai; Ling-Jun Ho; Chondroprotective effects and mechanisms of resveratrol in advanced glycation end products-stimulated chondrocytes. Arthritis Research & Therapy 2010, 12, R167-R167, 10.1186/ar3127.


