 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vsevolod V Gurevich | + 2793 word(s) | 2793 | 2021-02-08 07:46:45 | | | |
| 2 | Bruce Ren | -21 word(s) | 2772 | 2021-02-19 10:35:50 | | |
Video Upload Options
Arrestins are a small family of four proteins in most vertebrates that bind hundreds of different G protein-coupled receptors (GPCRs). Arrestin binding to a GPCR has at least three functions: precluding further receptor coupling to G proteins, facilitating receptor internalization, and initiating distinct arrestin-mediated signaling. The molecular mechanism of arrestin–GPCR interactions has been extensively studied and discussed from the “arrestin perspective”, focusing on the roles of arrestin elements in receptor binding.
1. Introduction
Arrestins are critical players in the homologous desensitization of G protein-coupled receptors (GPCRs). Active GPCRs interact with cognate heterotrimeric G proteins, catalyzing GDP/GTP exchange on their α-subunits. GTP binding to the G protein α-subunit promotes the dissociation of the G protein from the receptor and separation of its α- and βγ-subunits. The classical paradigm of homologous desensitization posits that eventually, the active receptor is phosphorylated by G protein-coupled receptor kinases (GRKs) (reviewed in [1]). Arrestins bind active phosphorylated receptors with high affinity [2]. The receptor binding of G proteins is transient due to the abundance of GTP in the cytoplasm, whereas the binding of arrestins to receptors is not. Thus, after receptor phosphorylation, arrestins outcompete G proteins, shutting down G protein-mediated signaling [3]. The formation of the arrestin-receptor complex also “activates” arrestins, inducing global conformational changes in the arrestin molecule that enable its transition into a state capable of binding the receptor with high affinity. “Active” GPCR-bound arrestins recruit numerous trafficking and signaling proteins [4], promoting receptor internalization and facilitating the signaling in several pathways [5][6]. The realm of arrestin activity goes beyond GPCRs and includes atypical seven transmembrane domain receptors (7TMRs), such as frizzled and smoothened receptors, receptor tyrosine kinases, cytokine receptors, and ion channels [7][8][9]. Quite a few reviews have discussed the role of particular arrestin elements in receptor binding and the consequent signaling [10], but the equally important GPCR side of the story has received a lot less attention.
Free and receptor-bound arrestins are different not only structurally [11] but also functionally [12]. Thus, it is important from a biological standpoint to determine what parts of the receptor facilitate arrestin’s transition from one state to the other, the molecular mechanisms whereby individual receptor elements facilitate this transition, and the role of particular interactions between receptor and arrestin residues in this process. Below, we summarize existing data and point out the gaps in current knowledge that need to be filled. We focus on GPCR elements that engage arrestins and, where known, on the actual role of these receptor elements in arrestin binding and its transition into an “active” signaling-competent conformation. We present fine molecular details which might be of interest only to those who work on the structure–function of arrestins and GPCRs. Therefore, we have emphasized the qualitative changes in both arrestins and GPCRs that contribute to the big picture of the regulation of cell signaling, where GPCRs, being the most numerous family of signaling proteins and targeted by about a third of clinically used drugs [13], play a prominent role. While sequence conservation in the GPCR super-family is fairly low [14], all GPCRs have a similar topology: an extracellular N-terminus, seven transmembrane α-helices (TM1-7) connected by three intracellular (ICL1-3) and three extracellular (ECL1-3) loops, and a cytoplasmic C-terminus, the beginning of which, between TM7 and the palmitoylation site, often forms helix 8.
2. Where Arrestins Start: Structure in the Basal State
Most vertebrates express four arrestin subtypes: visual arrestin-1 and -4 (We use systematic names of arrestin proteins, where the number after the dash indicates the order of cloning: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin-1), arrestin-3 (β-arrestin-2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin)), which are restricted to the photoreceptors in the retina where they quench light-induced signaling of the photopigments in rods and cones, and two ubiquitously expressed non-visual forms, arrestin-2 and -3 (also known as β-arrestin-1 and -2, respectively), which interact with hundreds of different GPCRs [15].
Comparison of the crystal structure of bovine arrestin-1 (PDB: 1CF1) [16], bovine arrestin-2 (PDB: 1G4M and 1G4R) [17], bovine arrestin-3 (PDB: 3P2D) [18][19], and tiger salamander (Ambystoma tigrinum) arrestin-4 (PDB: 1SUJ) [20] in the basal state reveals the overall similarity of these structures (Figure 1A). All arrestins consist of an N-domain and a C-domain, each formed by a “sandwich” consisting of two layers of β-strands. In addition to the extensive interface where the bodies of the two domains interact, there are two links between the domains: the inter-domain “hinge” and the C-tail. The length of the hinge was shown to be critical for GPCR binding in arrestin-1 [21], as well as non-visual arrestins 2 and 3 [22]. The C-tail makes a loop (not resolved in structures), after which it is anchored to the N-domain via the “three-element interaction” with β-strand I and the only α-helix in arrestins (Figure 1B). Several structural elements in visual arrestin-1 should be noted that might be important for the selectivity of visual arrestins for photopigments, in contrast to the much greater variety of GPCRs with which non-visual arrestins interact [20]. Bovine arrestin-1 contains valine in position 90. The large hydrophobic side chain of this valine is localized between the two layers of β-strands and apparently reduces the flexibility of the β-strand sandwich of the N-domain through interactions with several bulky hydrophobic partners. Valine in this position is conserved in arrestin-4 (also known as cone arrestin) but is replaced with serine or alanine in non-visual arrestins. While the N-domain of arrestin-4 shares similar H-bonding to that of arrestin-2, its C-domain structure resembles that of arrestin-1, making the structure of arrestin-4 a hybrid of non-visual arrestin-2 and visual arrestin-1. Notably, the loop between β-strands I and II in arrestin-1 contains R18, while the other three arrestins have proline in homologous positions [20]. It has been suggested that this additional positive charge in arrestin-1 ensures its greater preference for phosphorylated over unphosphorylated GPCRs [20]. Indeed, the difference in binding to the phosphorylated and unphosphorylated forms of the same receptor for both non-visual subtypes was experimentally shown to be much less dramatic than for arrestin-1 [23][24][25].
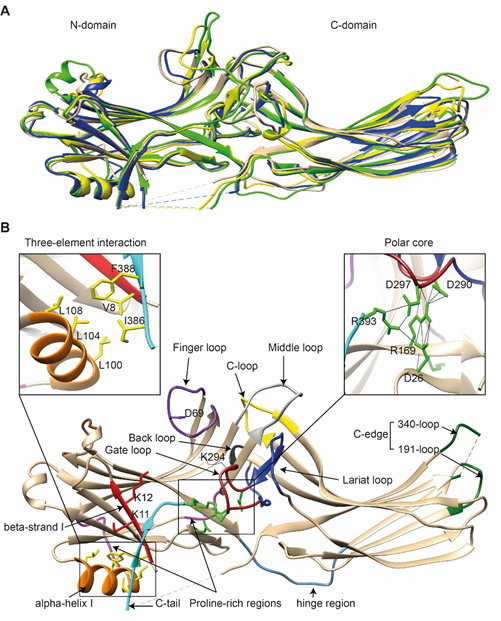
Figure 1. The basal structures of arrestins. (A) The crystal structures of bovine arrestin-1 (PDB: 1CF1, green [16]), bovine arrestin-2 (PDB: 1G4M, tan [17]), bovine arrestin-3 (PDB: 3P2D, blue [26]), and tiger salamander arrestin-4 (PDB: 1SUJ, yellow [20]) in the basal state are superimposed. Parts that are not resolved in the crystal structures are indicated by dashed lines. (B) Crystal structure of arrestin-2 (PDB: 1G4M [17]) in the basal state. Functionally important loops and critical residues are indicated and highlighted as follows: finger loop, purple; inter-domain hinge, blue-gray; β-strand I and the two lysines in it, red; α-helix I, orange; C-tail, light blue; hydrophobic side chains of residues in the α-helix, β-strand I, and β-strand XX of the C-tail mediating the three-element interaction, yellow; charged side chains of the five residues forming the polar core, green; polyproline motifs, light magenta; lariat loop, dark blue; its part called the gate loop, dark red; C-loop, yellow; back loop, black; C-edge loops, dark green. The side chain of the K294 in the gate loop pointing to the cavity of the N-domain is also shown. Close-up views of the three-element interaction and the polar core are shown in the left and right inserts, respectively.
Non-visual arrestins contain proline-rich regions (88–96 and 120–124 in arrestin-2, light magenta in Figure 1B), which are likely responsible for the binding of the SH3 domains of Src family kinases and other proteins containing SH3 domains through polyproline helix II (PPII). Proline-rich regions are absent in both visual arrestins. While the first segment (88–96) is unlikely to produce a PPII-type structure, residues 119–126, located in the connector between α-helix I and the body of the N-domain, can assume a PPII conformation, at least upon arrestin “activation” [27]. However, this requires rearrangement of P121 and N122 upon receptor binding [28]. Interestingly, while the connector between the α-helix and the N-domain (Figure 1B) in arrestin-2 has only one PPxP motif, the homologous element in arrestin-3 has two [27], which are consensus sequences for binding to the regulatory SH3 domains. In contrast to the other three subtypes, part of the receptor-binding surface in the C-domain of arrestin-3 (Q253-Q262) does not form a contiguous β-sheet via hydrogen bonding (as revealed by the crystal structure at 3.0-Å resolution), allowing increased flexibility of the receptor-binding side of the C-domain, which may explain the more promiscuous nature of arrestin-3, as compared to arrestin-2, in binding to different GPCRs.
So far, the highest resolution structure of any arrestin in its basal conformation is that of arrestin-2 (PDB: 1G4M; 1.9 Å). Therefore, we have used this structure for illustrative purposes (Figure 1B). Like all other subtypes, arrestin-2 contains a polar core (composed of five residues with charged side chains: D26, R169, D290, D297, and R393, green in Figure 1B; expanded in the right insert), which is one of the intra-molecular “clasps” that keep arrestins in their basal conformation. The lariat loop (N281-N311 in arrestin-2, dark blue in Figure 1B), which is highly conserved in all arrestins, contains the main counterion of R169 in the polar core, D290. Mutagenesis data suggest that the salt bridge between these two residues plays a central role in stabilizing the basal state [29][30][31][32]. The gate loop (part of the lariat loop, D290-N299 in arrestin-2; dark red in Figure 1B) supplies two out of the three negative charges in the polar core. Upon recruitment to the receptor, the polar core is destabilized, and the lariat loop, the C-tail (D383-R408 in arrestin-2, the D383-R393 part resolved in the crystal structure is shown in light blue in Figure 1B), and the N-domain undergo significant structural rearrangements [33][34][35]. The crystal structures of all arrestins in the basal conformation reveal a three-element interaction (Figure 1B, left insert) that involves bulky hydrophobic residues (side chains are shown in yellow in Figure 1B) in the α-helix (98–108 in arrestin-2, orange in Figure 1B) and β-strands I and XX of the N-domain and the C-tail, respectively [36]. The interaction of these three elements is disrupted upon binding to the receptor, resulting in structural rearrangement of β-strand I [37][38] and the release of the C-tail [38][39][40]. C-edge loops (dark green in Figure 1B) on the distal tip of the C-domain of arrestin-1 [41] and arrestin-2 were found in contact with the detergent or membrane in micelles or lipid nanodiscs, suggesting a role for these residues in the membrane anchoring of receptor-bound arrestins. This element in arrestin-1 was shown to fulfill this function upon rhodopsin binding [42].
3. Where Arrestins Go: The Structure of Receptor-Bound Arrestins
Arrestins undergo a global conformational rearrangement upon recruitment to the receptor. First, an interdomain twist, i.e., a rotation of the C-domain relative to the N-domain, is revealed by all structures of “active” arrestins. Interestingly, this twist was predicted long before the structures demonstrated it [43]. The extent of this twist varies between different structures of “active” arrestins [44][45]. “Active” arrestin conformations fall into two groups, those with small (~8°; PDB: 4ZRG, arrestin-1 R175E [46]; 3UGU, p44 splice variant of arrestin-1 [47]; 6K3F, C7pp-bound arrestin-3 [45]) and large (~18°, PDB: 5TV1, IP6-bound-arrestin-3; 4JQI, V2Rpp-bound arrestin-2 [48]; 4J2Q, arrestin-1 p44 [49]; 4ZWJ, rhodopsin-bound arrestin-1; 5W0P, rhodopsin-bound arrestin-1; 6UP7, NTS1R-bound arrestin-2) interdomain twists. Specific phosphorylation patterns, i.e., the number and spatial distribution of phosphates, have been suggested as a potential mechanism governing the extent of the interdomain twist [45], but this idea requires experimental testing. In addition to the extent of the interdomain twist, the orientation of arrestin-2 relative to the 7TM bundle of the receptor in complex with M2R [34], β1AR [50], and NTS1R [35] shows a 7°, 20°, and 90° difference, respectively, compared to the rhodopsin-arrestin-1 complex.
Second, the interaction between residues in the polar core is disrupted, as evidenced by the movement of D290 in the lariat loop away from R169, which is an essential contact stabilizing the basal conformation of arrestin-2 [35]. Disruption of the polar core upon activation is a shared phenomenon in the activation of all arrestin subtypes [48][49][50]. Third, the finger, lariat, middle, and C-loops undergo significant rearrangements. While the positions of the lariat and middle loops are similar in all reported receptor/arrestin complexes, the finger loop and C-loop adopt distinct conformations in different structures of “active” arrestins (Figure 2). Notably, the finger loop of V2Rpp-arrestin-2 complex forms an unstructured region superimposable neither with that of rhodopsin-arrestin-1 nor with that of β1AR-arrestin-2. The finger loop of arrestin-2 in complex with β1AR forms a β-hairpin, in contrast to the short α-helix in the rhodopsin-arrestin-1 or NTS1R-arrestin-2 complexes, and protrudes about 5 Å deeper into the interhelical cavity of the receptor [50].
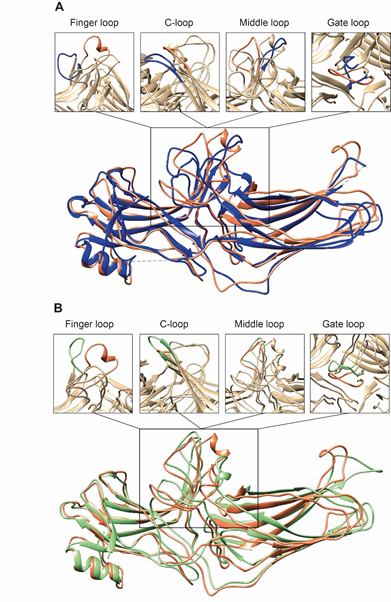
Figure 2. Comparison of arrestin-2 conformations in complex with M2R and NTS1R. (A) The crystal structures of arrestin-2 in the basal state (PDB: 1G4M, blue ) and in complex with NTS1R (PDB: 6UP7, orange ) are superimposed. (B) The crystal structures of arrestin-2 in complex with NTS1R (PDB: 6UP7, orange ) and in complex M2R (PDB: 6U1N, green ) are superimposed. The conformations of critical arrestin elements participating in receptor binding are compared in the close-up views above each panel. Specific regions are selected for comparison and the rest of structures are colored tan to highlight the difference in rearrangements upon activation.
4. Distinct Poses of Receptor-Bound Arrestin
The idea that receptor-bound arrestin does not necessarily have a fixed conformation but can assume different ones was proposed long ago [5]. The simplest explanation of the findings that arrestin binding to the same receptor phosphorylated by different GRKs, presumably at different sites, has distinct signaling consequences [51][52][53] is that the conformation of receptor-bound arrestin depends on the positions of the receptor-attached phosphates, and the actual conformation of bound arrestin determines its signaling capabilities, as was proposed more than a decade ago [54]. Indeed, mutations in the dopamine D1 receptor substituting particular phosphorylatable ICL3 residues with alanines or negatively charged phosphomimetics differentially affected its signaling to different protein kinases [55]. Moreover, some arrestin-3 residues significantly change its receptor preference [56][57], even though their homologues in arrestin-1 or -2 do not contact the bound receptors in any of the solved structures. This suggests that these elements participate in the binding, likely in alternative “flavors” of the complexes not resolved in structures. However, all this evidence is indirect. So far, there are very few pieces of direct evidence. First, the same arrestin-2 was found in strikingly different “poses” in complex with different GPCRs (Figure 3), indicating that there is more than one possible way of arrestin association with the receptor. Second, arrestins are capable of binding to the receptor intracellular core or only to the phosphorylated receptor C-terminus, leaving the core open for the binding of G proteins, forming megaplexes [58]. Third, as far as the complex of a single arrestin bound to a single receptor goes, distance measurements using the pulse electron paramagnetic resonance (EPR) technique called double electron–electron resonance between selected points in rhodopsin and bound arrestin-1 yielded more than one distance between each pair. While the most populated distances matched the crystal structure of the complex, to the delight of crystallographers, the presence of others suggested that the complex can have different “flavors”, only one of which was resolved in the crystal. Two experimental approaches can be used to prove this beyond a reasonable doubt. The first is the elucidation of the structures of arrestin complexes with the same receptor with phosphates in different positions (e.g., using mutant receptors with some of the phosphorylation sites eliminated). The second is the measurement of a sufficient number of distances between certain points in arrestin and in the receptor to develop models of the different flavors of the complex. Receptor and/or arrestin residues engaging the partner in a particular flavor, but not in others, can be mutated. If these mutations affect the observed binding (which is, by definition, the sum of all possible complexes), this would prove that those flavors exist and contribute to the measured interaction.
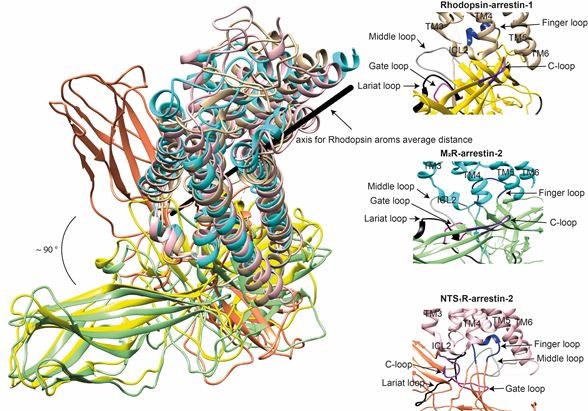
Figure 3. Comparison of the conformations of arrestin-1 and arrestin-2 in complex with different receptors. Left panel: The structures of the rhodopsin-arrestin-1 (PDB: 5W0P), M2R-arrestin-2 (PDB: 6U1N), and NTS1R-arrestin-2 (PDB: 6UP7) complexes are superimposed according to the best match in receptor sequences, which shows about a 90° difference in the position of arrestin-2 in complex with NTS1R as compared to the other structures. Right panels: Close-up views of the central crests of arrestins in contact with the receptor are shown for the rhodopsin (tan)-arrestin-1 (yellow) interface in the upper panel, the M2R (cyan)-arrestin-2 (green) interface in the middle panel, and the NTS1R (pink)-arrestin-2 (orange) interface in the lower panel. The finger (blue), middle (gray), lariat (black), gate (violet), and C- (purple) loops are highlighted to show the difference in the configurations of these elements in the structures.
References
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–46.
- Gurevich, V.V.; Gurevich, E.V. The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 2004, 25, 105–111, doi:10.1016/j.tips.2003.12.008.
- Carman, C.V.; Benovic, J.L. G-protein-coupled receptors: Turn-ons and turn-offs. Curr. Opin. Neurobiol. 1998, 8, 335–344.
- Xiao, K.; McClatchy, D.B.; Shukla, A.K.; Zhao, Y.; Chen, M.; Shenoy, S.K.; Yates, J.R.; Lefkowitz, R.J. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 12011–12016.
- Gurevich, E.V.; Gurevich, V.V. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006, 7, 236.
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297.
- Lefkowitz, R.J.; Rajagopal, K.; Whalen, E.J. New roles for beta-arrestins in cell signaling: Not just for seven-transmembrane receptors. Mol. Cell 2006, 24, 643–652, doi:10.1016/j.molcel.2006.11.007.
- Khedmat, S.; Seyedabadi, M.; Ghahremani, M.H.; Ostad, S.N. Cyclooxygenase 2 plays a role in Emdogain-induced proliferation. J. Periodontal Res. 2011, 46, 67–73, doi:10.1111/j.1600-0765.2010.01313.x.
- Seyedabadi, M.; Rahimian, R.; Ghia, J.E. The role of alpha7 nicotinic acetylcholine receptors in inflammatory bowel disease: Involvement of different cellular pathways. Expert Opin. Ther. Targets 2018, 22, 161–176, doi:10.1080/14728222.2018.1420166.
- Gurevich, V.V.; Gurevich, E.V. Custom-designed proteins as novel therapeutic tools? The case of arrestins. Expert Rev. Mol. Med. 2010, 12, e13.
- Chen, Q.; Iverson, T.M.; Gurevich, V.V. Structural Basis of Arrestin-Dependent Signal Transduction. Trends Biochem. Sci. 2018, 43, 412–423, doi:10.1016/j.tibs.2018.03.005.
- Gurevich, V.V.; Gurevich, E.V. The new face of active receptor bound arrestin attracts new partners. Structure 2003, 11, 1037–1042.
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842.
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272.
- Indrischek, H.; Prohaska, S.J.; Gurevich, V.V.; Gurevich, E.V.; Stadler, P.F. Uncovering missing pieces: Duplication and deletion history of arrestins in deuterostomes. BMC Evol. Biol. 2017, 17, 163.
- Hirsch, J.A.; Schubert, C.; Gurevich, V.V.; Sigler, P.B. The 2.8 A crystal structure of visual arrestin: A model for arrestin’s regulation. Cell 1999, 97, 257–269.
- Han, M.; Gurevich, V.V.; Vishnivetskiy, S.A.; Sigler, P.B.; Schubert, C. Crystal structure of beta-arrestin at 1.9 A: Possible mechanism of receptor binding and membrane translocation. Structure 2001, 9, 869–880.
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol. 2011 Feb 25;406(3):467-78
- Zhan, X.; Stoy, H.; Kaoud, T.S.; Perry, N.A.; Chen, Q.; Perez, A.; Els-Heindl, S.; Slagis, J.V.; Iverson, T.M.; Beck-Sickinger, A.G.; et al. Peptide mini-scaffold facilitates JNK3 activation in cells. Sci. Rep. 2016, 6, 21025.
- Sutton, R.B.; Vishnivetskiy, S.A.; Robert, J.; Hanson, S.M.; Raman, D.; Knox, B.E.; Kono, M.; Navarro, J.; Gurevich, V.V. Crystal Structure of Cone Arrestin at 2.3Å: Evolution of Receptor Specificity. J. Mol. Biol. 2005, 354, 1069–1080, doi:10.1016/j.jmb.2005.10.023.
- Vishnivetskiy, S.A.; Hirsch, J.A.; Velez, M.-G.; Gurevich, Y.V.; Gurevich, V.V. Transition of arrestin in the active receptor-binding state requires an extended interdomain hinge. J. Biol. Chem. 2002, 277, 43961–43968.
- Hanson, S.M.; Cleghorn, W.M.; Francis, D.J.; Vishnivetskiy, S.A.; Raman, D.; Song, X.; Nair, K.S.; Slepak, V.Z.; Klug, C.S.; Gurevich, V.V. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J. Mol. Biol. 2007, 368, 375–387.
- Kovoor, A.; Celver, J.; Abdryashitov, R.I.; Chavkin, C.; Gurevich, V.V. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 1999, 274, 6831–6834.
- Celver, J.; Vishnivetskiy, S.A.; Chavkin, C.; Gurevich, V.V. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 2002, 277, 9043–9048.
- Gimenez, L.E.; Kook, S.; Vishnivetskiy, S.A.; Ahmed, M.R.; Gurevich, E.V.; Gurevich, V.V. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. J. Biol. Chem. 2012, 287, 9028–9040, doi:10.1074/jbc.M111.311803.
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 2011, 406, 467–478, doi:10.1016/j.jmb.2010.12.034.
- Chen, Q.; Perry, N.A.; Vishnivetskiy, S.A.; Berndt, S.; Gilbert, N.C.; Zhuo, Y.; Singh, P.K.; Tholen, J.; Ohi, M.D.; Gurevich, E.V.; et al. Structural basis of arrestin-3 activation and signaling. Nat. Commun. 2017, 8, 1427.
- Hirsch, J.A.; Schubert, C.; Gurevich, V.V.; Sigler, P.B. A Model for Arrestin’s Regulation: The 2.8 Å Crystal Structure of Visual Arrestin. Cell 1999, 97, 257–269, doi:10.1016/S0092-8674(00)80735-7.
- Vishnivetskiy, S.A.; Paz, C.L.; Schubert, C.; Hirsch, J.A.; Sigler, P.B.; Gurevich, V.V. How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 1999, 274, 11451–11454.
- Gurevich, V.V.; Pals-Rylaarsdam, R.; Benovic, J.L.; Hosey, M.M.; Onorato, J.J. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J. Biol. Chem. 1997, 272, 28849–28852.
- Pan, L.; Gurevich, E.V.; Gurevich, V.V. The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J. Biol. Chem. 2003, 278, 11623–11632.
- Vishnivetskiy, S.A.; Zheng, C.; May, M.B.; Karnam, P.C.; Gurevich, E.V.; Gurevich, V.V. Lysine in the lariat loop of arrestins does not serve as phosphate sensor. J. Neurochem. 2020, doi:10.1111/jnc.15110.
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 2019, 29, 971–983, doi:10.1038/s41422-019-0256-2.
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302.
- Huang, W.; Masureel, M.; Qianhui, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308, doi:10.1038/s41586-020-1953-1.
- Milano, S.K.; Pace, H.C.; Kim, Y.M.; Brenner, C.; Benovic, J.L. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 2002, 41, 3321–3328.
- Vishnivetskiy, S.A.; Schubert, C.; Climaco, G.C.; Gurevich, Y.V.; Velez, M.-G.; Gurevich, V.V. An additional phosphate-binding element in arrestin molecule: Implications for the mechanism of arrestin activation. J. Biol. Chem. 2000, 275, 41049–41057.
- Zhuo, Y.; Vishnivetskiy, S.A.; Zhan, X.; Gurevich, V.V.; Klug, C.S. Identification of receptor binding-induced conformational changes in non-visual arrestins. J. Biol. Chem. 2014, 289, 20991–21002, doi:10.1074/jbc.M114.560680.
- Vishnivetskiy, S.A.; Francis, D.J.; Van Eps, N.; Kim, M.; Hanson, S.M.; Klug, C.S.; Hubbell, W.L.; Gurevich, V.V. The role of arrestin alpha-helix I in receptor binding. J. Mol. Biol. 2010, 395, 42–54.
- Hanson, S.M.; Francis, D.J.; Vishnivetskiy, S.A.; Kolobova, E.A.; Hubbell, W.L.; Klug, C.S.; Gurevich, V.V. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. USA 2006, 103, 4900–4905.
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature 2015, 523, 561–567.
- Lally, C.C.; Bauer, B.; Selent, J.; Sommer, M.E. C-edge loops of arrestin function as a membrane anchor. Nat. Commun. 2017, 8, 14258.
- Modzelewska, A.; Filipek, S.; Palczewski, K.; Park, P.S. Arrestin interaction with rhodopsin: Conceptual models. Cell Biochem. Biophys. 2006, 46, 1–15.
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G protein-Coupled Receptors. Cell 2017, 170, 457–469, doi:10.1016/j.cell.2017.07.002.
- Min, K.; Yoon, H.J.; Park, J.Y.; Baidya, M.; Dwivedi-Agnihotri, H.; Maharana, J.; Chaturvedi, M.; Chung, K.Y.; Shukla, A.K.; Lee, H.H. Crystal Structure of β-Arrestin 2 in Complex with CXCR7 Phosphopeptide. Structure 2020, 28, 1014–1023.e4, doi:10.1016/j.str.2020.06.002.
- Granzin, J.; Stadler, A.; Cousin, A.; Schlesinger, R.; Batra-Safferling, R. Structural evidence for the role of polar core residue Arg175 in arrestin activation. Sci. Rep. 2015, 5, 15808, doi:10.1038/srep15808.
- Granzin, J.; Cousin, A.; Weirauch, M.; Schlesinger, R.; Büldt, G.; Batra-Safferling, R. Crystal structure of p44, a constitutively active splice variant of visual arrestin. J. Mol. Biol. 2012, 416, 611–618, doi:10.1016/j.jmb.2012.01.028.
- Shukla, A.K.; Manglik, A.; Kruse, A.C.; Xiao, K.; Reis, R.I.; Tseng, W.C.; Staus, D.P.; Hilger, D.; Uysal, S.; Huang, L.Y.; et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 2013, 497, 137–141, doi:10.1038/nature12120.
- Kim, Y.J.; Hofmann, K.P.; Ernst, O.P.; Scheerer, P.; Choe, H.W.; Sommer, M.E. Crystal structure of pre-activated arrestin p44. Nature 2013, 497, 142–146, doi:10.1038/nature12133.
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β(1)-adrenoceptor. Nature 2020, 583, 862–866, doi:10.1038/s41586-020-2419-1.
- Kim, J.; Ahn, S.; Ren, X.-R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc. Nat. Acad. Sci. USA 2005, 102, 1442–1447, doi:10.1073/pnas.0409532102.
- Ren, X.R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Nat. Acad. Sci. USA 2005, 102, 1448–1453, doi:10.1073/pnas.0409534102.
- Choi, M.; Staus, D.P.; Wingler, L.M.; Ahn, S.; Pani, B.; Capel, W.D.; Lefkowitz, R.J. G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Sci. Signal. 2018, 11, eaar7084.
- Tobin, A.B.; Butcher, A.J.; Kong, K.C. Location, location, location...site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 2008, 29, 413–420, doi:10.1016/j.tips.2008.05.006.
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Nat. Acad. Sci. USA 2020, 117, 14139–14149.
- Gimenez, L.E.; Vishnivetskiy, S.A.; Baameur, F.; Gurevich, V.V. Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J. Biol. Chem. 2012, 287, 29495–29505, doi:10.1074/jbc.M112.366674.
- Gimenez, L.E.; Babilon, S.; Wanka, L.; Beck-Sickinger, A.G.; Gurevich, V.V. Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell. Signal. 2014, 26, 1523–1531, doi:10.1016/j.cellsig.2014.03.019.
- Nguyen, A.H.; Thomsen, A.R.B.; Cahill, T.J.; Huang, R.; Huang, L.-Y.; Marcink, T.; Clarke, O.B.; Heissel, S.; Masoudi, A.; Ben-Hail, D.; et al. Structure of an endosomal signaling GPCR–G protein–β-arrestin megacomplex. Nat. Struct. Mol. Biol. 2019, 26, 1123–1131, doi:10.1038/s41594-019-0330-y.


