 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Muhammad S Soyfoo | + 3241 word(s) | 3241 | 2021-02-08 04:48:26 | | | |
| 2 | Peter Tang | -10 word(s) | 3231 | 2021-02-24 12:33:34 | | |
Video Upload Options
Sjogren’s syndrome (SS) is a chronic autoimmune disease characterized by the infiltration of exocrine glands including salivary and lachrymal glands responsible for the classical dry eyes and mouth symptoms (sicca syndrome). The spectrum of disease manifestations stretches beyond the classical sicca syndrome with systemic manifestations including arthritis, interstitial lung involvement, and neurological involvement. The pathophysiology underlying SS is not well deciphered, but several converging lines of evidence have supported the conjuncture of different factors interplaying together to foster the initiation and perpetuation of the disease
1. Sjögren’s Syndrome
Sjögren’s syndrome (SS) is one of the most common autoimmune rheumatic diseases. SS is characterized by the immune-mediated destruction of exocrine glands, including lachrymal and salivary glands (SGs). Two types of SS have been defined: Primary SS (pSS), which occurs in the absence of other autoimmune diseases, and secondary SS (sSS), which is associated with other autoimmune disorders such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and scleroderma [1][2]. SS is characterized by a high sex preponderance with a ratio of nine female for one male. This sexual imbalance suggests an involvement of estrogens and androgens in the development of the pathology [3][4] that could account for an incidence increase of pSS during the post-menopausal stage, at the age of 40–60 years old [5]. In general, the diagnosis is based on the combination of several oral and ocular sicca symptoms, the presence of the autoimmune manifestations such the production of autoantibodies anti-Ro/SSA, the labial biopsy showing a focal lymphocytic infiltration (focus score ≥ 1 per 4 mm2) [6]. The pathophysiology of SS is very complex, multifactorial, and consecutive to several genetic, hormonal, environmental, and immunological risk factors. Due to its complexity, the clinical course of the pathology can be divided in several phases: An initiation phase consecutive to endogenous and exogenous factors, a dysregulation of salivary glands epithelial cells (SGECs), and an immune system activation and chronicity of inflammation induced by B cells hyperactivity [7]. The combination of all these events culminates in the destruction of the salivary gland architecture, and development of keratoconjunctivitis sicca and xerostomia. Each phase plays a significant role in the disease. The transition from the innate immune system to the adaptive system responses and the variety of cell types involved could explain the difficulties in developing an efficient therapeutic strategy for pSS.
2. Innate Immune Cells Involved in Sjögren’s Syndrome
A growing body of evidence indicates that innate immunity plays a crucial role in the pathogenesis of pSS, especially in the initiation and progression towards autoimmunity [8]. We will discuss the role of each cell type implicated in this process often called autoimmune epithelitis.
2.1. Dendritic Cells
Dendritic cells (DCs) are professional antigen presenting cells. They act as sentinels capturing and processing antigens, migrating in T cell areas to initiate immunity and differentiating in response to a variety of stimuli such as Toll-like receptor (TLR) ligands, cytokines, innate lymphocytes, and immune complexes [9]. DCs play a key role in pSS as they display an aberrant phenotype causing them to accumulate in SGs [10][11][12]. Saliva from pSS patients is characterized by an upregulation of C-C chemokine receptor type 5 (CCR5) and CCR5 ligands such as CC chemokine ligand type 3 (CCL3) and type 4 (CCL4) that play an important role for the effective migration of DCs to inflamed tissues. In addition, lower numbers of blood DCs in patients with pSS may be consecutive to the aberrant regulation of apoptosis [13].
Plasmacytoid DCs (pDCs) are a specific subset of DCs that can be activated by self-antigens through TLR-7 and TRL-9 [14][15] and to a lesser extent TLR-2, TRL-4, and TRL-9 [16], leading to the production of type I interferon (IFN). Type I IFN acts through autocrine and paracrine circuits sustaining a continuous reinforcing inflammatory loop. It also induces the production of the B cell activating factor (BAFF) by monocyte circulating cells and DCs contributing to the activation and differentiation of B cells into plasma cells secreting antibodies [16][17].
Follicular DCs (fDCs) originate from fibroblast precursor cells and play an essential part in the structure of ectopic germinal centers. FDCs promote B cells survival and proliferation in the long run by retaining on their surface immune-complexes (IC), formed by antigen-antibody-complement. Contrary to other DCs, fDCs do not display phagocytic activity and lack lysosomes and lysozyme [18][19].
2.2. Macrophages
Macrophages are the main tissue resident leucocytes and are characterized by pleomorphic phenotypes. According to their microenvironment, they can display pro-inflammatory or anti-inflammatory activities, immunogenic or tolerogenic activities, and tissue destructive or tissue regenerative activities [20][21].
In SGs specimens from patients with pSS, macrophages tend to appear early and their number is positively correlated with the biopsy focus score [22]. Macrophages are activated by interferon gamma (IFN-γ) and interleukin (IL)-17 secreted by type 1 T helper cells (Th1) and type 17 T helper cells (Th17), respectively [23]. Activated macrophages produce inflammatory cytokines such as IL-1, tumor necrosis factor alpha (TNFα), IL-18, and metalloproteases (MMPs) leading to epithelial cell damage [24][25]. Activated macrophages can also act as antigenic peptide presenting cells through their major histocompatibility complex class II (MHC-II) and interact with antigen-specific CD4+ T cells [23]. The latter, once activated, evolves in autoreactive clones that may perpetuate the activation of macrophages, themselves sustaining a pro-inflammatory auto-maintained loop [25].
Manoussakis et al., studied MSG biopsies from pSS patients and demonstrated increased infiltration by macrophages together with a marked expression of IL-18 by infiltrating macrophages. Moreover, IL-18 levels correlated with lymphoma risk factors such as persistent C4 hypocomplementemia and SG enlargement [26].
Beside SGs, this process may affect other epithelia such as the eye epithelium leading to the development of squamous metaplasia which represents the end stage of ocular involvement in pSS patients [27][28].
2.3. Mast Cells
Mast cells are immune cells mainly found in connective tissues. Their role in allergy and anaphylaxis is well established. However, a great deal of evidence underlines their possible involvement in tissue healing, angiogenesis, and autoimmune exacerbation [29].
In pSS patients, Leehan et al., confirmed that fibrosis of minor salivary glands (MSG) is a pathological feature of pSS that positively correlates witha focus score and is not age-related [30]. Another study identified that mast cells are strongly associated with the fibrosis and fatty infiltration of SGs. It is hypothesized that they promote fibrosis through interaction with local fibroblasts and through the production of enzymes cleaving and activating MMPs, which are essential mediators of tissue injury [31].
Mast cells express TLR-2 and TLR-4, as well as receptors for IL-1 including interleukin-1 receptor type 1 (IL1R1) and suppressor of tumorigenicity 2 (ST2). Mast cells activation through TLR-2 and TLR4 lead to the production of IL-1, TNF-α, IL-33, and chemokines such as C-X-C motif chemokine ligand 1 (CXCL1) and C-X-C motif chemokine ligand 2 (CXCL2), which recruit neutrophilic granulocytes and DCs. The activation of mast cells through IL1R1 and ST2 allows them to interact with T and B cells, interfering with antibody production [29]. The activation of ST2 on mast cells through IL-33 leads to the production of pro-inflammatory cytokines such as IL-1, IL-6, IL-13 [32], and induces a TH2 polarization of CD4+ T cells.
2.4. Salivary Gland Epithelial Cells (SGECs)
Salivary gland epithelial cells (SGECs) form the acinar secretory structure and the ductal excretory structures in SGs [33]. SGECs constitute the main target of auto-immunity in pSS, described as an autoimmune epithelitis [34]. Over recent years, it has become clear that SGECs also fulfill an important role in the initiation of autoimmunity.
In pSS patients, the loss of polarity of SGECs plays a crucial part in the onset of the local inflammatory process. Indeed, a decrease in occludin and zonula occludens 1 (ZO-1) expression and a redistribution of claudin to the basolateral plasma membrane have been observed in SGs from pSS patients. Furthermore, the exposure of isolated SGs cells from healthy controls to pro-inflammatory cytokines such as TNF-α and IFN-γ reproduced the alterations observed in pSS patients [35][36]. By altering the tight junction integrity of SGECs, the local cytokine production may therefore account for the secretory gland dysfunction observed in pSS patients, and subsequent decrease in saliva quality and quantity [35].
In genetically susceptible subjects, environmental stimuli such as viruses may trigger salivary gland epithelial cells (SGECs) through TLR activation [37][38]. More precisely, the activation of TLR-2 and TLR-4 expressed on the surface of SGECs results in the expression of mediators of immune activation (such as the intercellular adhesion molecule 1 (ICAM-1)), CD40, and major histocompatibility complex 1 (MHC-1) [39], as well as in IL-15 secretion inducing the proliferation of activated B and T cells and the generation and maintenance of natural killer (NK) cells. Beside leading to the expression of ICAM-1, CD40, and MHC-1, the activation of endosomal TLR3 leads to the secretion of BAFF that promotes the activation and maturation of B cells. TLR3 activation also contributes to SGEC anoikis, a form of apoptosis that is triggered by a loss of cell attachment to the extracellular matrix (ECM), thereby releasing exosomes and apoptotic blebs containing autoantigens such as Ro/SSA and La/SSB that drive autoimmunity in pSS by attracting both classical DC and pDC within SGs [40][41][42].
Activated SGECs produce chemokines that attract immune cells and contribute to the formation of germinal centers, including C-X-C motif chemokine ligand 9 (CXCL-9), C-X-C motif chemokine ligand 10 (CXCL-10), C-X-C motif chemokine ligand 12 (CXCL12), C-X-C motif chemokine ligand 13 (CXCL13) and C-C chemokine ligand 19 (CCL19), and C-C chemokine ligand 21 (CCL21) [43][44]. An increased epithelial production of cytokines such as IL-1, IL-6, and TNFα may also contribute to create a pro-inflammatory environment [45].
Activated SGECs develop the ability to act as non-professional antigen-presenting cells [46] by expressing co-stimulation molecules (CD80 and CD86) [47] and MHC-I (HLA-ABC) and MHC-II (HLA-DR), adhesion molecules such as ICAM1, vascular cell adhesion molecule 1 (VCAM-1) [46][48]. Thus, SGECs appear suitably equipped for the presentation of antigenic peptides and the transmission of activation signals to T cells [44][49].
2.5. Endothelial Cells
Endothelial cells, expressing CD31, form a one-cell thick walled layer called endothelium that upholster blood and lymphatic vessels. Beside bringing immunes cells to inflammation sites, endothelial cells take an active and regulatory role in inflammatory processes [50]. In response to IL-1 and TNFα, activated endothelium express adhesion molecules such as ICAM-1, VCAM-1, and E- and P-selectins that allow the interaction and migration of blood immune cells to inflamed tissues [51].
In pSS patients, the expression of ICAM-1 positively correlates with a focus score of salivary biopsies [52]. Both strong vascular endothelial growth factor C (VEGF-C) and vascular endothelial growth factor receptor 3 (VEGFR-3) expression were reported in MSGs from pSS patients [53]. As a result, increased and anatomically aberrant lymphatic neovascularization leads to a persistent extravasation of immune cells. In another study, defective lymphatic vessels were also characterized by the overproduction of CCL-21 that increased the infiltration of immune cells into inflamed tissues [54].
2.6. Mucosal-Associated Invariant T (MAIT) Cells
Mucosal-associated invariant T (MAIT) cells are innate-like T cells and can therefore be considered a bridge between innate and adaptive responses [55]. MAIT cells express an invariant T cell receptor (TCR) α-chain (Vα7.2–Jα33 in humans) and CD161 which is typically expressed by NK cells. They recognize vitamin B-related peptides through the evolutionary conserved non-polymorphic MHC-I-related molecule (MR1) [56]. In response to different stimuli, MAIT cells also have the capacity to express both CD4 and/or CD8 co-receptors. MAIT cells are characterized by a natural memory function and by their capacity to rapidly produce Th1, type 2 helper cells (Th2), and Th17 cytokines [55][57].
Very little is known about the contribution of MAIT cells in the pathogenesis of pSS. Wang et al. found that MAIT cells are decreased in peripheral blood circulation but are increased in SGs from pSS patients compared to healthy controls. From a functional point of view, MAIT cells from pSS patients were mainly CD4+ and naïve, in contrast with MAIT cells from controls that were almost exclusively CD8+. In addition, MAIT cells of pSS patients displayed lower levels of activation with a reduced expression of CD69 and CD154, and lower levels of TNFα and IFN-γ. The aberrant phenotype of MAIT cells in pSS patients may lead to the dysregulation of the local immune responses, which would trigger local damage in SGs and auto-immunity [57].
2.7. Natural Killer (NK) Cells
Natural Killer (NK) cells are a cytolytic component of the innate immune system. They have the ability to sense the pathological changes of self-cells and therefore take an important part in the immune surveillance of tumor cells and virus-infected cells [58]. NK cells express the NKp30 receptor that is recognized by DCs and lead to the production of Th1 cytokines such as IFN-γ and IL-12 [59].
NK cells are enriched in MSGs from pSS patients and their presence correlate with the focus score [60]. In addition, NK cells overexpress the NKp30 receptor and SGECs express B7-H6, the ligand for NKp30. Taken together, this may explain the hyperactivity of NK cells and the interrelation with SGECs and DCs that lead to a subsequent activation of innate and adaptive immunity. The expression of B7-H6 by SGECs may also be involved in the homing of NK cells in SGs [60]. Another study identified a subset of NK cells that expresses NKp44 and produces IL-22 in SGs from pSS patients. This subgroup has however been reclassified and is now part of Innate Lymphoid Cells (ILCs) [61].
2.8. Natural Killer T (NKT) Cells
NK T (NKT) cells are immune components that share features of both T cells and NK cells. They discriminate self from non-self-antigens and produce prompt immune responses against Gram-negative bacteria [62]. They are a major source of IL-4 and to a lesser extent of IFN [63]. Invariant NKT (iNKT) cells are a special subset of NKT cells that seems to play a pivotal role in the regulation of immunity. They express CD161 (typical of NK cells) and a semi-invariant T cell receptor (TCR). By linking CD1d in B cells with their invariant TCR, iNKT cells are able to suppress B cell auto-reactivity. In addition, under certain circumstances, they can express both CD8+ and CD4+, which leads to the production of Th1 and Th2 cytokines [62][64].
A decreased number of NKT cells was observed in the peripheral blood of pSS patients, which could be explained by apoptosis or homing in SGs [65]. Another study reported an increased number of iNKT in peripheral blood but a complete absence of iNKT cells together with an increased number of auto-reactive B cells in SGs from pSS patients [66]. These data were corroborated by showing that the lack of CD1d following B-cells hyperactivation lead to a greater release of autoantibodies [67]. In spite of the studies supporting the candidacy of NK cells in the SG of pSS patients, there is actually no sufficient proof bolstering their role as participating actively in the pathology of SS.
2.9. Innate Lymphoid Cells (ILCs)
Innate lymphoid cells (ILCs) are the innate counterparts of T helper lymphocytes [68][69]. They are mostly concentrated at epithelial barriers and rapidly release cytokines in response to environmental triggers. They can be classified into three subsets according to the expression of specific transcription factors and the production of cytokines that mirror the subsets of helper T. ILC1 express the T-Box Transcription Factor 21 (TBX21, also named T-bet), produce IFN-γ, and respond to intracellular pathogens such as viruses. ILC2 express the transcription factor GATA Binding Protein 3 (GATA-3), produce IL-4, IL-5 and IL-13, and respond to extracellular parasites and allergens. ILC3 express the transcription factor Retinoic acid-related orphan receptor gamma t (RORγt), produce IL-17A and IL-22, and react to extracellular pathogens such as bacteria and fungi [68][70][71].
ILCs have been identified in the SGs from pSS patients [61][72] and may contribute to the formation of germinal center-like structures [73]. Blokland et al. showed that the presence of ILC1 was associated with a higher disease activity score (ESSDAI). ILC1 could contribute to the pathogenesis of pSS through the massive production of IFN-γ, but the underlying mechanisms remain largely elusive [74]. In addition, an increased IFN signature and reduced frequencies of ILC2 and ILC3 was associated with a high expression of Fas cell surface death receptor (Fas also named CD95) by ILC2 and ILC3. It was hypothesized that the increased Fas expression on ILC2 and ILC3 may induce an apoptosis of these cells. These observations corroborate previous studies in mice that reported a link between type I IFN, pDC activation, and apoptosis of circulating ILC2 and ILC3 [75][76][77].
A subset of ILC3 that was originally classified as NK cells because of its expression of NKp44 was identified in SGs from pSS patients. It was found to be a major source of IL-22 together with Th17 cells. The frequency of ILC3 was positively correlated to the focus score [61]. Additional studies are needed to further evaluate the ILC3 function in SGs from pSS patients. Currently, their role in the pathogenesis of SS remains to be determined and detailed. There is not enough data purporting their role as being active players in SS pathology.
Figure 1 summarizes the action of the different players of innate immunity in the pathogenesis of pSS.
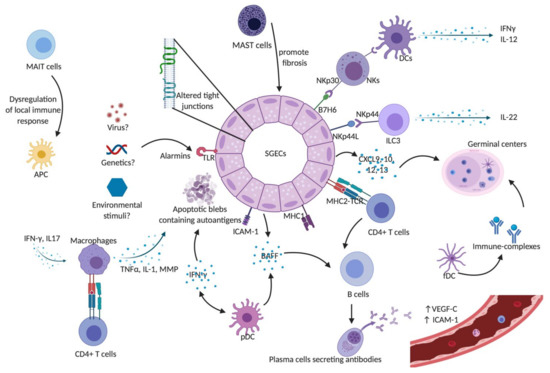
Figure 1. Innate immunity in Sjögren’s syndrome. SGECs constitute the main target of auto-immunity in pSS, described as an autoimmune epithelitis. SGECs exhibit a subverted architecture mainly characterized by altered tight junctions. In genetic susceptible subjects, environmental stimuli such as viruses may trigger salivary gland epithelial cells (SGECs) through TLR activation. Activated SGECs secrete the BAFF that promotes activation and maturation of B cells. SGECs also produce chemokines such as CXCR9, 10, 11, and 12 that attract immune cells and contribute to the formation of germinal centers. Activated SGECs have the ability to act as non-professional antigen-presenting cells by expressing MHC-I, (HLA-ABC) and MHC-II (HLA-DR), adhesion molecules such as ICAM1 allowing them to activate T cells. TLR activation also contributes to SGEC apoptosis, releasing autoantigens that drive autoimmunity in pSS. Activated macrophages produce inflammatory cytokines such as IL-1, TNFα, and MMPs leading to epithelial cell damage. They can also act as antigenic peptide presenting cells through their MHC-II and interact with antigen-specific CD4+ T cells. pDCs lead to the production of type I IFN that acts through autocrine and paracrine circuits feeding a continuous reinforcing inflammatory loop. It also induces the production of BAFF, production contributing to the activation of B cells into plasma cells. fDCs play an essential part in the structure of ectopic germinal centers and retain on their surface immune-complexes, formed by antigen-antibody-complement. Mast cells contribute to the fibrosis and fatty infiltration of salivary glands (SGs). The aberrant phenotype of MAIT cells in pSS patients may lead to the dysregulation of the local immune responses, which would trigger local damage in SGs and auto-immunity. NK cells express the NKp30 receptor that is recognized by DCs and lead to the production of Th1 cytokines such as IFN-γ and IL-12. SGECs express B7-H6, the ligand for NKp30. Taken together, this may explain the hyperactivity of NK cells and the cross-talk with SGECs and DCs that lead to a subsequent activation of innate and adaptive immunity. A subset of ILC3 was found to be a major source of IL-22 in SGECs. Abbreviations: APC: Antigen presenting cells; BAFF: B-cell activating factor; CXCL9: C-X-C motif chemokine type 9; CXCL10: C-X-C motif chemokine type 10; CXCL12: C-X-C motif chemokine type 12; CXCL13: C-X-C motif chemokine type 13; DCs: dendritic cells; fDCs: follicular dendritic cells; ICAM-1: intercellular adhesion molecule 1; IFN-γ: interferon gamma; IL-: interleukin; ILC3: innate Lymphoid Cells type 3 MAIT: Mucosal-associated invariant T cells; MHC-I: major histocompatibility complex class I; MHC-II: major histocompatibility complex class II; MMPs: metalloproteases; NK: natural killer cells; NKp44L: NKp44 ligand; pDCs: plasmacytoid dendritic cells; SGECs: salivary glands epithelial cells; TCR: T cell receptor; TLR: Toll like receptor; TNFα: tumor necrosis factor alpha; VEGF-C: vascular endothelial growth factor C.
References
- Fox, R.I.; Kang, H.I. Pathogenesis of Sjogren’s syndrome. Rheum. Dis. Clin. N. Am. 1992, 18, 517–538.
- Fox, R.I.; Michelson, P. Approaches to the treatment of Sjogren’s syndrome. J. Rheumatol. Suppl. 2000, 61, 15–21.
- Basbaum, A.I. Opioid regulation of nociceptive and neuropathic pain. Clin. Neuropharmacol. 1992, 15 Pt A (Suppl. S1), 372A.
- Sullivan, D.A. Sex hormones and Sjogren’s syndrome. J. Rheumatol. Suppl. 1997, 50, 17–32.
- Nguyen, C.Q.; Peck, A.B. Unraveling the pathophysiology of Sjogren syndrome-associated dry eye disease. Ocul. Surf. 2009, 7, 11–27.
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.M.; Alexander, E.L.; Carsons, S.E.; Daniels, T.E.; Fox, P.C.; Fox, R.I.; Kassan, S.S.; et al. Classification criteria for Sjogren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum Dis. 2002, 61, 554–558.
- Parisis, D.; Chivasso, C.; Perret, J.; Soyfoo, M.S.; Delporte, C. Current State of Knowledge on Primary Sjogren’s Syndrome, an Autoimmune Exocrinopathy. J. Clin. Med. 2020, 9, 2299.
- Kiripolsky, J.; McCabe, L.G.; Kramer, J.M. Innate immunity in Sjogren’s syndrome. Clin. Immunol. 2017, 182, 4–13.
- Steinman, R.M.; Hemmi, H. Dendritic cells: Translating innate to adaptive immunity. Curr. Top. Microbiol. Immunol. 2006, 311, 17–58.
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775.
- Ozaki, Y.; Ito, T.; Son, Y.; Amuro, H.; Shimamoto, K.; Sugimoto, H.; Katashiba, Y.; Ogata, M.; Miyamoto, R.; Murakami, N.; et al. Decrease of blood dendritic cells and increase of tissue-infiltrating dendritic cells are involved in the induction of Sjogren’s syndrome but not in the maintenance. Clin. Exp. Immunol. 2010, 159, 315–326.
- Wildenberg, M.E.; Welzen-Coppens, J.M.; van Helden-Meeuwsen, C.G.; Bootsma, H.; Vissink, A.; van Rooijen, N.; van de Merwe, J.P.; Drexhage, H.A.; Versnel, M.A. Increased frequency of CD16+ monocytes and the presence of activated dendritic cells in salivary glands in primary Sjogren syndrome. Ann. Rheum. Dis. 2009, 68, 420–426.
- Hillen, M.R.; Pandit, A.; Blokland, S.L.M.; Hartgring, S.A.Y.; Bekker, C.P.J.; van der Heijden, E.H.M.; Servaas, N.H.; Rossato, M.; Kruize, A.A.; van Roon, J.A.G.; et al. Plasmacytoid DCs From Patients With Sjogren’s Syndrome Are Transcriptionally Primed for Enhanced Pro-inflammatory Cytokine Production. Front. Immunol. 2019, 10, 2096.
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hanninen, A.; Nordstrom, D.C. Activation of plasmacytoid dendritic cells by apoptotic particles-mechanism for the loss of immunological tolerance in Sjogren’s syndrome. Clin. Exp. Immunol. 2018, 191, 301–310.
- Swiecki, M.; Colonna, M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 2010, 234, 142–162.
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485.
- Vakaloglou, K.M.; Mavragani, C.P. Activation of the type I interferon pathway in primary Sjogren’s syndrome: An update. Curr. Opin. Rheumatol. 2011, 23, 459–464.
- Aloisi, F.; Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217.
- Frank, G.B.; Kokate, T.G. Blockade of high K+ contractures and Ca(+)+-dependent slow action potentials in frog skeletal muscle by opioid drugs. Prog. Clin. Biol. Res. 1990, 328, 283–286.
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349.
- Morell, M.; Varela, N.; Maranon, C. Myeloid Populations in Systemic Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2017, 53, 198–218.
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the minor salivary gland infiltrates in Sjogren’s syndrome. J. Autoimmun. 2010, 34, 400–407.
- Mills, K.H. Induction, function and regulation of IL-17-producing T cells. Eur. J. Immunol. 2008, 38, 2636–2649.
- Gliozzi, M.; Greenwell-Wild, T.; Jin, W.; Moutsopoulos, N.M.; Kapsogeorgou, E.; Moutsopoulos, H.M.; Wahl, S.M. A link between interferon and augmented plasmin generation in exocrine gland damage in Sjogren’s syndrome. J. Autoimmun. 2013, 40, 122–133.
- Rizzo, C.; Grasso, G.; Destro Castaniti, G.M.; Ciccia, F.; Guggino, G. Primary Sjogren Syndrome: Focus on Innate Immune Cells and Inflammation. Vaccines 2020, 8, 272.
- Manoussakis, M.N.; Boiu, S.; Korkolopoulou, P.; Kapsogeorgou, E.K.; Kavantzas, N.; Ziakas, P.; Patsouris, E.; Moutsopoulos, H.M. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: Correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007, 56, 3977–3988.
- Kinoshita, S.; Nakamura, T.; Nishida, K. Pathological keratinization of ocular surface epithelium. Adv. Exp. Med. Biol. 2002, 506 Pt. A, 641–646.
- McNamara, N.A. Molecular mechanisms of keratinizing ocular surface disease. Optom Vis. Sci. 2010, 87, 233–238.
- Conti, P.; Stellin, L.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G. Advances in Mast Cell Activation by IL-1 and IL-33 in Sjogren’s Syndrome: Promising Inhibitory Effect of IL-37. Int. J. Mol. Sci. 2020, 21, 4297.
- Leehan, K.M.; Pezant, N.P.; Rasmussen, A.; Grundahl, K.; Moore, J.S.; Radfar, L.; Lewis, D.M.; Stone, D.U.; Lessard, C.J.; Rhodus, N.L.; et al. Minor salivary gland fibrosis in Sjogren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S112), 80–88.
- Skopouli, F.N.; Li, L.; Boumba, D.; Stefanaki, S.; Hanel, K.; Moutsopoulos, H.M.; Krilis, S.A. Association of mast cells with fibrosis and fatty infiltration in the minor salivary glands of patients with Sjogren’s syndrome. Clin. Exp. Rheumatol. 1998, 16, 63–65.
- Xu, D.; Jiang, H.R.; Kewin, P.; Li, Y.; Mu, R.; Fraser, A.R.; Pitman, N.; Kurowska-Stolarska, M.; McKenzie, A.N.; McInnes, I.B.; et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10913–10918.
- de Paula, F.; Teshima, T.H.N.; Hsieh, R.; Souza, M.M.; Nico, M.M.S.; Lourenco, S.V. Overview of Human Salivary Glands: Highlights of Morphology and Developing Processes. Anat. Rec. 2017, 300, 1180–1188.
- Manoussakis, M.N.; Moutsopoulos, H.M. Sjogren’s syndrome: Autoimmune epithelitis. Baillieres Best Pract Res. Clin. Rheumatol. 2000, 14, 73–95.
- Ewert, P.; Aguilera, S.; Alliende, C.; Kwon, Y.J.; Albornoz, A.; Molina, C.; Urzua, U.; Quest, A.F.; Olea, N.; Perez, P.; et al. Disruption of tight junction structure in salivary glands from Sjogren’s syndrome patients is linked to proinflammatory cytokine exposure. Arthritis Rheum. 2010, 62, 1280–1289.
- Barrera, M.J.; Bahamondes, V.; Sepulveda, D.; Quest, A.F.; Castro, I.; Cortes, J.; Aguilera, S.; Urzua, U.; Molina, C.; Perez, P.; et al. Sjogren’s syndrome and the epithelial target: A comprehensive review. J. Autoimmun. 2013, 42, 7–18.
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of intrinsic epithelial activation in the pathogenesis of Sjogren’s syndrome. J. Autoimmun. 2010, 35, 219–224.
- Goules, A.V.; Kapsogeorgou, E.K.; Tzioufas, A.G. Insight into pathogenesis of Sjogren’s syndrome: Dissection on autoimmune infiltrates and epithelial cells. Clin. Immunol. 2017, 182, 30–40.
- Hashimoto, A.; Nishikawa, T.; Hayashi, T.; Fujii, N.; Harada, K.; Oka, T.; Takahashi, K. The presence of free D-serine in rat brain. FEBS Lett. 1992, 296, 33–36.
- Manoussakis, M.N.; Spachidou, M.P.; Maratheftis, C.I. Salivary epithelial cells from Sjogren’s syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. J. Autoimmun. 2010, 35, 212–218.
- Kiripolsky, J.; Kramer, J.M. Current and Emerging Evidence for Toll-Like Receptor Activation in Sjogren’s Syndrome. J. Immunol. Res. 2018, 2018, 1246818.
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Gourzi, V.C.; Konsta, O.D.; Baltatzis, G.E.; Tzioufas, A.G. Toll-like receptor 3 stimulation promotes Ro52/TRIM21 synthesis and nuclear redistribution in salivary gland epithelial cells, partially via type I interferon pathway. Clin. Exp. Immunol. 2014, 178, 548–560.
- Ogawa, N.; Ping, L.; Zhenjun, L.; Takada, Y.; Sugai, S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjogren’s syndrome. Arthritis Rheum. 2002, 46, 2730–2741.
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of epithelial cells in the pathogenesis of Sjogren’s syndrome. Clin. Rev. Allergy Immunol. 2007, 32, 225–230.
- Boumba, D.; Skopouli, F.N.; Moutsopoulos, H.M. Cytokine mRNA expression in the labial salivary gland tissues from patients with primary Sjogren’s syndrome. Br. J. Rheumatol. 1995, 34, 326–333.
- Tsunawaki, S.; Nakamura, S.; Ohyama, Y.; Sasaki, M.; Ikebe-Hiroki, A.; Hiraki, A.; Kadena, T.; Kawamura, E.; Kumamaru, W.; Shinohara, M.; et al. Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjogren’s syndrome. J. Rheumatol. 2002, 29, 1884–1896.
- Matsumura, R.; Umemiya, K.; Goto, T.; Nakazawa, T.; Kagami, M.; Tomioka, H.; Tanabe, E.; Sugiyama, T.; Sueishi, M. Glandular and extraglandular expression of costimulatory molecules in patients with Sjogren’s syndrome. Ann. Rheum Dis. 2001, 60, 473–482.
- Kapsogeorgou, E.K.; Dimitriou, I.D.; Abu-Helu, R.F.; Moutsopoulos, H.M.; Manoussakis, M.N. Activation of epithelial and myoepithelial cells in the salivary glands of patients with Sjogren’s syndrome: High expression of intercellular adhesion molecule-1 (ICAM.1) in biopsy specimens and cultured cells. Clin. Exp. Immunol. 2001, 124, 126–133.
- Mitsias, D.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. The role of epithelial cells in the initiation and perpetuation of autoimmune lesions: Lessons from Sjogren’s syndrome (autoimmune epithelitis). Lupus 2006, 15, 255–261.
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815.
- Mikulowska-Mennis, A.; Xu, B.; Berberian, J.M.; Michie, S.A. Lymphocyte migration to inflamed lacrimal glands is mediated by vascular cell adhesion molecule-1/alpha(4)beta(1) integrin, peripheral node addressin/l-selectin, and lymphocyte function-associated antigen-1 adhesion pathways. Am. J. Pathol. 2001, 159, 671–681.
- Turkcapar, N.; Sak, S.D.; Saatci, M.; Duman, M.; Olmez, U. Vasculitis and expression of vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and E-selectin in salivary glands of patients with Sjogren’s syndrome. J. Rheumatol. 2005, 32, 1063–1070.
- Alunno, A.; Ibba-Manneschi, L.; Bistoni, O.; Rosa, I.; Caterbi, S.; Gerli, R.; Manetti, M. Mobilization of lymphatic endothelial precursor cells and lymphatic neovascularization in primary Sjogren’s syndrome. J. Cell Mol. Med. 2016, 20, 613–622.
- Gunn, M.D.; Tangemann, K.; Tam, C.; Cyster, J.G.; Rosen, S.D.; Williams, L.T. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 258–263.
- Toubal, A.; Nel, I.; Lotersztajn, S.; Lehuen, A. Mucosal-associated invariant T cells and disease. Nat. Rev. Immunol. 2019, 19, 643–657.
- Gold, M.C.; McLaren, J.E.; Reistetter, J.A.; Smyk-Pearson, S.; Ladell, K.; Swarbrick, G.M.; Yu, Y.Y.; Hansen, T.H.; Lund, O.; Nielsen, M.; et al. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J. Exp. Med. 2014, 211, 1601–1610.
- Wang, J.J.; Macardle, C.; Weedon, H.; Beroukas, D.; Banovic, T. Mucosal-associated invariant T cells are reduced and functionally immature in the peripheral blood of primary Sjogren’s syndrome patients. Eur. J. Immunol. 2016, 46, 2444–2453.
- Izumi, Y.; Ida, H.; Huang, M.; Iwanaga, N.; Tanaka, F.; Aratake, K.; Arima, K.; Tamai, M.; Kamachi, M.; Nakamura, H.; et al. Characterization of peripheral natural killer cells in primary Sjogren’s syndrome: Impaired NK cell activity and low NK cell number. J. Lab. Clin. Med. 2006, 147, 242–249.
- Ferlazzo, G.; Tsang, M.L.; Moretta, L.; Melioli, G.; Steinman, R.M.; Munz, C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J. Exp. Med. 2002, 195, 343–351.
- Rusakiewicz, S.; Nocturne, G.; Lazure, T.; Semeraro, M.; Flament, C.; Caillat-Zucman, S.; Sene, D.; Delahaye, N.; Vivier, E.; Chaba, K.; et al. NCR3/NKp30 contributes to pathogenesis in primary Sjogren’s syndrome. Sci. Transl. Med. 2013, 5, 195ra96.
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F.; Campisi, G.; Alessandro, R.; Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann. Rheum Dis. 2012, 71, 295–301.
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336.
- Yoshimoto, T. The Hunt for the Source of Primary Interleukin-4: How We Discovered That Natural Killer T Cells and Basophils Determine T Helper Type 2 Cell Differentiation In Vivo. Front. Immunol. 2018, 9, 716.
- Rizzo, C.; La Barbera, L.; Lo Pizzo, M.; Ciccia, F.; Sireci, G.; Guggino, G. Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome. Int. J. Mol. Sci. 2019, 20, 5435.
- Sudzius, G.; Mieliauskaite, D.; Siaurys, A.; Viliene, R.; Butrimiene, I.; Characiejus, D.; Dumalakiene, I. Distribution of Peripheral Lymphocyte Populations in Primary Sjogren’s Syndrome Patients. J. Immunol. Res. 2015, 2015, 854706.
- Guggino, G.; Ciccia, F.; Raimondo, S.; Giardina, G.; Alessandro, R.; Dieli, F.; Sireci, G.; Triolo, G. Invariant NKT cells are expanded in peripheral blood but are undetectable in salivary glands of patients with primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2016, 34, 25–31.
- Yang, J.Q.; Wen, X.; Kim, P.J.; Singh, R.R. Invariant NKT cells inhibit autoreactive B cells in a contact- and CD1d-dependent manner. J. Immunol. 2011, 186, 1512–1520.
- Zook, E.C.; Kee, B.L. Development of innate lymphoid cells. Nat. Immunol. 2016, 17, 775–782.
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066.
- Ebbo, M.; Crinier, A.; Vely, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678.
- Shikhagaie, M.M.; Germar, K.; Bal, S.M.; Ros, X.R.; Spits, H. Innate lymphoid cells in autoimmunity: Emerging regulators in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 164–173.
- Wenink, M.H.; Leijten, E.F.A.; Cupedo, T.; Radstake, T. Review: Innate Lymphoid Cells: Sparking Inflammatory Rheumatic Disease? Arthritis Rheumatol. 2017, 69, 885–897.
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462.
- Blokland, S.L.M.; van den Hoogen, L.L.; Leijten, E.F.A.; Hartgring, S.A.Y.; Fritsch, R.; Kruize, A.A.; van Roon, J.A.G.; Radstake, T. Increased expression of Fas on group 2 and 3 innate lymphoid cells is associated with an interferon signature in systemic lupus erythematosus and Sjogren’s syndrome. Rheumatology 2019, 58, 1740–1745.
- Maazi, H.; Banie, H.; Aleman Muench, G.R.; Patel, N.; Wang, B.; Sankaranarayanan, I.; Bhargava, V.; Sato, T.; Lewis, G.; Cesaroni, M.; et al. Activated plasmacytoid dendritic cells regulate type 2 innate lymphoid cell-mediated airway hyperreactivity. J. Allergy Clin. Immunol. 2018, 141, 893–905 e6.
- Zhang, Z.; Cheng, L.; Zhao, J.; Li, G.; Zhang, L.; Chen, W.; Nie, W. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J. Clin. Invest. 2015, 125, 3692–3703.
- Duerr, C.U.; McCarthy, C.D.; Mindt, B.C.; Rubio, M.; Meli, A.P.; Pothlichet, J.; Eva, M.M.; Gauchat, J.F.; Qureshi, S.T.; Mazer, B.D.; et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat. Immunol. 2016, 17, 65–75.


