 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Danai Barlampa | + 3367 word(s) | 3367 | 2021-02-09 12:17:04 | | | |
| 2 | Vivi Li | Meta information modification | 3367 | 2021-02-18 08:56:40 | | |
Video Upload Options
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder among women of reproductive age. It is a heterogeneous condition characterized by reproductive, endocrine, metabolic, and psychiatric abnormalities. More than one pathogenic mechanism is involved in its development. On the other hand, the hypothalamus plays a crucial role in many important functions of the body, including weight balance, food intake, and reproduction. A high-fat diet with a large amount of long-chain saturated fatty acids can induce inflammation in the hypothalamus. Hypothalamic neurons can sense extracellular glucose concentrations and participate, with a feedback mechanism, in the regulation of whole-body glucose homeostasis. When consumed nutrients are rich in fat and sugar, and these regulatory mechanisms can trigger inflammatory pathways resulting in hypothalamic inflammation. The latter has been correlated with metabolic diseases, obesity, and depression.
1. Introduction
The hypothalamus, being part of diencephalon, is situated beneath the thalamus and above the midbrain. It consists of many nuclei, with different functional roles, organized in four regions: the preoptic, anterior (supraoptic), median (tuberal), and posterior (mammillary) hypothalamus, each of which has a lateral, medial, and periventricular zone [1] (Figure 1A). The hypothalamus regulates genetically preprogrammed behaviors, such as temperature, feeding, defensive and aggressive behaviors, as well as emotional responses to stress and panic-related behavior, libido, and reproduction [2]. Inflammatory pathways can be activated in the hypothalamus. The preoptic area is known to control thermoregulation, reproduction, and electrolyte balance. In the median region, dorsomedial, ventromedial, and arcuate (infundibular) nuclei are involved in regulation of body-weight balance, food intake, satiety, thirst, and circadian rhythms. Dysfunction of these nuclei causes hyperphagia and obesity [2]. Gonadotropin-releasing hormone (GnRH) neurons are a heterogeneous population of hypothalamic neurons, which control reproduction. The majority of GnRH neuronal cell bodies are located in the arcuate nucleus (part of the mediobasal hypothalamus) and in the medial preoptic area. GnRH is secreted in a pulsatile fashion and, through the portal circulation, stimulates synthesis and secretion of luteinizing hormone (LH) and follicle stimulating hormone (FSH) from the anterior pituitary gland. These gonadotropins are responsible for the secretion of gonadal steroids, which exert a negative feedback in the brain [3].
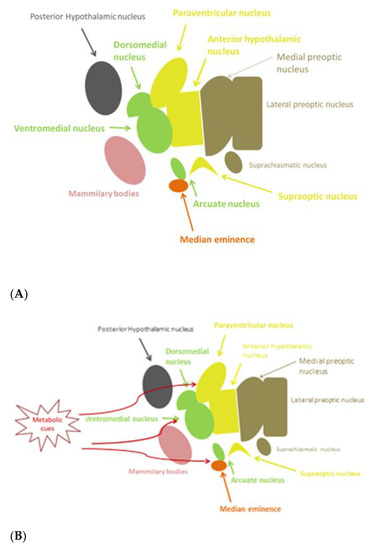
Figure 1. (A) Anatomy of hypothalamic nuclei; (B) Areas affected by metabolic cues in the hypothalamus.
Hypothalamic inflammation bears characteristics of chronic low-grade inflammation, notably at the molecular level [4]. On the one hand, a high-fat diet (HFD) has been associated to hypothalamic inflammation [5]. Over-nutrition leads rapidly to an inflammatory response in the hypothalamus and to a disrupted appetite [6]. In rodents, only 24–72 h following initiation of HFD, an elevation of markers of hypothalamic inflammation is observed [7]. It has been noticed that hypothalamic inflammation occurs earlier than excess weight gain and obesity do. Eventually, hypothalamic inflammation can be both the cause and the outcome of a diet-induced metabolic disease. In addition, known metabolic disorders are associated with inflammation in the peripheral organs [4]. On the other hand, diet-induced obesity induces a chronic low-grade inflammatory response in the body and is one of the most representative metabolic disorders. Over-nutrition with a high-caloric diet results in hyperplasia and hypertrophy of adipocytes. Thus, these cells lead to adipose tissue dysfunction and ectopic fat accumulation [8]. Ectopic fat storage in the liver, pancreas, skeletal muscle, and visceral adipose tissue contributes to the development of insulin resistance and inflammation in these target organs [9]. Inflammation in the pancreatic islets seen in type 2 diabetes mellitus (T2DM) includes pancreatic infiltration by immune cells, such as macrophages [10]. The excess of unsaturated fatty acids (FAs), such as palmitic acid, stimulates pancreatic islet inflammation as well [11]. Then, β-cell dysfunction and insulin resistance ensue.
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder among reproductive-aged women. Its prevalence ranges from 6–20% depending on the used diagnostic criteria [12]. The Rotterdam diagnostic criteria for PCOS are the most frequently used. According to them, the PCOS definition should include two of the following three criteria: 1. clinical or/and biochemical hyperandrogenism; 2. chronic anovulation; and 3. polycystic ovarian morphology, after the exclusion of other secondary causes [13]. PCOS is a complex condition characterized not only by reproductive but also metabolic, endocrine, and psychiatric features. Patients with PCOS present reproductive (such as subfertility and infertility) and metabolic abnormalities (such as marked insulin resistance combined with elevated risk for T2DM, cardiovascular disease, hepatic steatosis, dyslipidemia, and obesity) [14]. Depression and anxiety complete the profile of this heterogeneous condition. Studies have shown that these metabolic disturbances are associated with inflammation in peripheral target organs [15]. More than one pathophysiologic mechanism is involved in the development of PCOS, including environmental, genetic, and epigenetic mechanisms. A defect in the GnRH/gonadotropins neuro-endocrine axis is also one of the potential causative mechanisms of PCOS [16]. The generation of GnRH pulses is driven by a dynamic balance between excitatory and inhibitory signals. The kisspeptinergic system is one of them and serves as regulator of GnRH pulsatility [17]. The energy status of the human body (either positive or negative) affects the kisspeptinergic system. Therefore, the metabolic elements from the periphery interfere actively with the centers of reproduction in the brain [17]. In PCOS women, insulin resistance and the concomitant hyperinsulinemia result in hyperandrogenemia thus participating in the pathogenic mechanism of PCOS [18].
2. Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS Physiopathologic Mechanisms
2.1. Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS
PCOS is a highly prevalent endocrine disorder affecting a woman’s overall health long term. It is a complex condition with a broad spectrum of clinical manifestations and associated morbidities [19]. Women with PCOS have an increased risk for developing cardiovascular disease, metabolic syndrome, reproductive abnormalities, depression, and certain forms of cancer. A combination of clinical symptoms, such as menstrual abnormalities, acne, hirsutism, and alopecia, is reported in PCOS [20]. In a systematic review and meta-analysis, the authors reported that women with PCOS present with abnormal eating disorders three times more frequently as compared to women without PCOS [21]. The prevalence of bulimia nervosa and binge eating disorder were increased in these women. Of note, 50% of individuals with a lifetime diagnosis of bulimia nervosa and 32% with binge eating disorder have also experienced major depression, while almost 12% of those with bulimia nervosa or binge eating disorder present with generalized anxiety disorder. Indeed, 27–50% of women with PCOS report depressive symptoms compared to 19% of women without PCOS. In another study, in PCOS women, the overall prevalence of psychiatric morbidity was 50%, while specifically that of anxiety disorder was 38.6% [20].
Women with PCOS present higher androgen and estradiol (E2) concentrations, hyperinsulinemia, and suppressed sex hormone binding globulin (SHBG) concentrations compared with women without PCOS, while women with PCOS and obesity more often present ovarian dysfunction and amenorrhea [22].
Obesity is a common symptom (50% of PCOS women are overweight). By 40 years of age, up to 40% of women with PCOS will develop impaired glucose tolerance while 8–10% of PCOS patients are diagnosed with T2DM [14][23]. Women with PCOS present higher concentrations of triglycerides and low-density lipoprotein cholesterol (LDL-C) and lower levels of high-density lipoprotein cholesterol (HDL-C) than those of controls [24][25].
2.2. Physiopathologic Mechanisms in PCOS
A variety of physiopathologic mechanisms are involved in the pathogenesis and development of PCOS. However, the exact pathophysiological mechanism is still unknown. GnRH neurons are the neuronal denominator of a complex neuronal network, which regulates the reproductive system. A defect in the pulsatile secretion of GnRH and a subsequent elevation of LH circulating concentrations, which, in turn, enhance ovarian androgen secretion, seems to be involved in the pathophysiology of this pathologic entity [26]. Due to their anatomical position, GnRH neurons interact with a range of neuroendocrine and metabolic inputs [27]. Kisspeptin-neurokinin B and dynorphin neurons, known as KNDγ neurons, represent major regulators of GnRH pulsatility [17]. These are sited predominantly in an area corresponding to the posterior arcuate nucleus within the mediobasal hypothalamus. By classical lesion studies, this area was identified as the putative location for the GnRH pulse generator in primates [17]. GnRH neurons express the Gpr54 gene (G protein-coupled receptor GPR54), which encodes for the kisspeptin receptor, suggesting the involvement of kisspeptin in the regulation of GnRH secretion [28]. Specifically, kisspeptin and neurokinin B stimulate GnRH pulsatility while dynorphin inhibits it. Kisspeptin is considered a potent stimulator of the HPO axis [17].
Insulin resistance (predominantly in the liver, adipose tissue, and muscles) is the most prevalent metabolic perturbation in women with PCOS, affecting 65–70% of all patients [29]. It is followed by compensatory hyperinsulinemia and has been related to the reproductive defects of PCOS [18]. Hyperinsulinemia contributes to androgen-depended anovulation through distinct mechanisms. Insulin enhances ovarian androgen biosynthesis via its receptors in the theca cells [30]. The ovaries, in PCOS, do not demonstrate insulin resistance [31]. In addition, insulin increases the frequency and amplitude of GnRH pulsatility (via upregulation of GnRH gene expression in GnRH neurons) and subsequently LH pulsatile secretion. These effects contribute, in turn, to hyperandrogenemia (HA) [26]. The latter stimulates further GnRH pulse frequency by inhibiting the progesterone negative feedback, which finally leads to an increase of LH secretion [26].
The human body has to ensure a sufficient energy reservoir for reproduction. This requires a dynamic interaction between peripheral and central tissues [32]. The role of the kisspeptinergic system is to recognize the energy status of the body and to translate this information into the brain by enhancing the secretion of GnRH [17]. In detail, fasting and calorie restriction suppresses LH pulse frequency and increases LH pulse amplitude [32]. Leptin is a hormone secreted from adipose tissue and plays a crucial role in reproduction and in the regulation of LH release. In recent studies, leptin has been suggested to stimulate directly (via its own receptors) and/or indirectly kisspeptin neurons [33]. In states of low leptin activity, such as in mutations of leptin or its receptors and in functional hypothalamic amenorrhea, the release of GnRH and gonadotropins is impaired. [17][34]. The hypothalamic kisspeptin system is sensitive to changes in the body energy status and may be altered in conditions of persistent negative energy balance. However, it remains to be defined whether metabolic stress associated with an excess of energy stores, such as obesity, might have an impact on this system as well. There are controversial studies regarding the effect of diet-induced obesity on the expression of the kisspeptin mRNA (Kiss1) gene. Persistent obesity in DBA/2J mice, predisposed to reproductive disorders, is associated with marked suppression of Kiss1 mRNA in the arcuate and anteroventral periventricular nuclei [35]. However, following initiation of HFD in male Srague-Dawley rats, an increase in the expression of the Kiss1 gene in the hypothalamus has been demonstrated [36]. These findings suggest that Kiss1 neurons in both the arcuate and the anteroventral periventricular nuclei may be targets for metabolic regulation. Further research is needed regarding the exact effect of negative energy balance and metabolic stress regarding the responsiveness of the GnRH/gonadotropin system to the stimulatory effects of kisspeptin.
3. Hypothalamic Inflammation as a Potential Pathophysiological Mechanism of the Heterogeneity of PCOS
3.1. Hypothalamic Inflammation-Induced Metabolic Disorders
Several studies show that hypothalamic inflammation participates in the development and pathogenesis of metabolic diseases, such as obesity, diabetes, metabolic syndrome, hypertension, and dyslipidemia, mainly by affecting the homeostasis of food intake, energy balance, insulin and leptin signaling, and glucose metabolism in liver and fatty acid oxidation [37][38].
Key to this neuroendocrine regulation of energy balance is the melanocortin system. In particular, in a healthy state, the arcuate nucleus contains the orexigenic AgRP/neuropeptide Y (NPY) and the anorexigenic POMC/cocaine- and amphetamine- regulated transcript (CART) neurons [39][40]. Leptin and insulin, which are secreted by the adipose tissue and pancreas, respectively, cross the blood brain barrier and bind to their receptors in the arcuate nucleus. The blood vessels of this circumventricular organ are fenestrated and lack tight junctions, allowing exposure to solutes from the circulation [41]. In a fed state, leptin binds to the leptin receptor ObR/LrpR and increases POMC gene expression, thus releasing the active form of α-melanocyte-stimulating hormone (α-MSH) [42]. Then, α-MSH binds to the melanocortin 3/4 (MC3/4R) receptor in the PVN but also in the dorsomedial hypothalamic nucleus, in the lateral hypothalamus, as well as in the ventromedial hypothalamus, exerting its anorectic effects [43][44]. In contrast, in fasting state, insulin activates the AgRP promoter and represses the POMC promoter, resulting in an increase in food intake [45]. AgRP is an inverse agonist of the MC3/4R receptor and counters the anorectic effects of α-MSH [46].
As already mentioned, studies in mice showed that hypothalamic inflammation presents within few hours after initiation of HFD and especially prior to peripheral inflammation and notable weigh gain [5][7]. Since the first days, a local inflammation is noticed, in particular an activation of microglia mainly in the mediobasal hypothalamus in the arcuate nucleus, the anterior part of PVN, and in the median eminence [43] (Figure 1B). A study in mice showed the need for an adequate presence of fats and carbohydrates in the diet in order for microglia to expand. Feeding mice with a very high-fat very low-carbohydrate diet did not affect microglia cell proliferation [47]. The latter disrupts neuronal regulation of the energy balance by inducing inflammation, thus resulting in degradation of POMC neurons [48]. This inflammation promotes stress of the endoplasmic reticulum, leading to insulin and leptin resistance in the central nervous system (CNS) [43]. The increased number of activated microglia possibly contributes further to neurodegeneration [47].
Τhe destruction mainly of the PVN and ventromedial hypothalamic nuclei following inflammation leads to overeating, increased weight gain, obesity, and T2DM. The activation of several inflammatory pathways is known to induce this local inflammation. The activation of the proinflammatory JNK1 pathway in AgRP neurons increases spontaneous firing in these cells and, along with central leptin resistance, it leads to hyperphagia, weight gain, and adiposity [49]. In addition, activation of the inhibitory inflammatory IKK2 pathway mitigates the AgRP response to insulin and impairs glucose homeostasis. In another study, overactivation of the STAT/SOCS3 intracellular pathway by increased concentrations of leptin, resulting from growing adipose tissue, not only leads to leptin but also to insulin resistance at the level of CNS [50]. Further to this, mitochondria are also essential for energy regeneration and sensing of energy cellular demands via production of ROS [51]. Following HFD, impairment of the balance between production and disposal of ROS in the hypothalamus can result in endoplasmic reticulum stress, leptin resistance, and changes in eating behavior and in glucose utilization. All this inevitably leads to obesity and T2DM [48]. Similarly, production of transforming growth factor β (TGFβ) by hypothalamic astrocytes following HFD induces RNA stress, especially in POMC neurons, which affects proper function of proteins, thus leading to dysfunction of POMC [52]. An important outcome of this process is that the hypothalamus can no longer exert appropriate control on hepatic gluconeogenesis [53]. As a result, an early defect in a mechanism tightly connected to hypothalamic neuronal protein function can contribute to glucose intolerance via alteration of the hepatic physiology [53][54]. Furthermore, when autophagy in AgRP neurons is inhibited, mice eat less and are lean, whereas when autophagy is inhibited in POMC neurons, mice eat more and are obese [55][56][57]. Following HFD, autophagy activity in the mediobasal hypothalamus is impaired severely. The consequent hypothalamic inflammation further inhibits hypothalamic autophagy in lean mice, thus inducing increased caloric intake and obesity [58][59] (Figure 2).
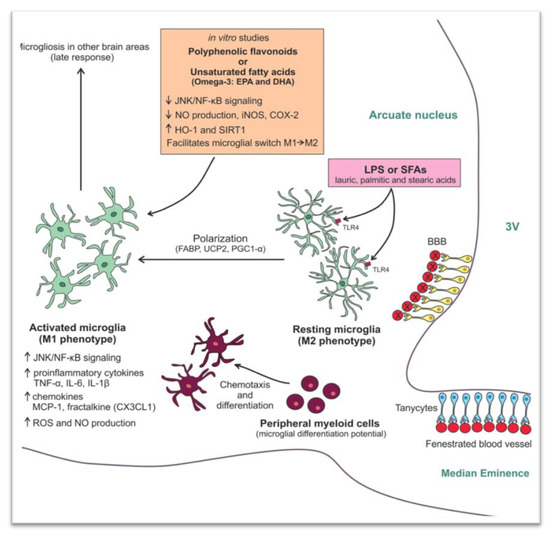
Figure 2. Mechanisms involved in the high fat diet (HFD) -induced hypothalamic microgliosis at molecular level [60]. Reproduced with permission from Licio A. Velloso, Hypothalamic Microglial Activation in Obesity: A Mini-Review; published by frontiers in Neuroscience, 2018.
Another potential mechanism for the development of peripheral insulin resistance and T2DM is through alteration of the activity of autonomic nerves, which innervate the liver, muscles, adipose tissue, and pancreas [61]. The PVN nuclei exert their regulatory influence via several mechanisms, including modulation of the activity of the sympathetic nervous system. In addition, the reduced hypothalamic insulin and glucose sensitivity along with the subsequently altered neuroendocrine regulatory mechanisms (including HPA axis activation) may also participate in the development of insulin resistance [62].
Insulin also controls fatty acids (FA) release from white adipose tissue (WAT) through direct effects on adipocytes and indirectly through hypothalamic signaling by reducing sympathetic nervous system outflow to WAT. Uncontrolled FA release from WAT promotes lipotoxicity, which is characterized by inflammation and insulin resistance. In support, after the initiation of a three-day HFD to Sprague-Dawley rats, a 37% increase in caloric intake and elevated base-line free FAs and insulin levels compared with controls rats were observed. The results were that overfeeding did not impair insulin signaling in WAT, but it abolished the ability of mediobasal hypothalamus insulin to suppress WAT lipolysis and hepatic glucose production. Insulin levels in Sprague-Dawley rats were moderately raised during the fed period, consistent with systemic insulin resistance [63]. Dyslipidemia could also follow the same pathophysiological pathway. However, there are still no human studies to support direct hypothalamic inflammation-induced dyslipidemia in PCOS.
3.2. Potential Hypothalamic Inflammation-Associated Mechanisms in Reproductive Disorders
Women with PCOS and obesity tend to have more notable endocrine disturbances [22]. They present more severe cycle disturbances and they need increased dosages of ovulation induction drugs compared to lean PCOS patients, while they demonstrate increased risk of non-response to such treatment [64]. It has been shown by several studies that acute inflammation affects reproduction. Administration of cytokines or lipopolysaccharide (LPS) intracerebroventricularly in female rats represses GnRH and LH gene expression and reduces GnRH neuropeptide release and gonadotropin concentrations [65]. In any case, obesity causes low-grade inflammation in the organism, but its effects on reproduction via GnRH neurons are to be investigated. Several studies have shown that HFD negatively affects the estrous cycle as it is associated with a high incidence of anovulation. However, it is still unknown if this is what happens in PCOS [66]. Studies in animals have shown that a high-fat and high-sugar (HFHS) diet resulted in irregularity of the estrous cycle and anovulation. Regarding follicular development, animals on the HFHS diet had significantly elevated ovarian cyst counts compared to controls [67]. In another study, advanced glycation end-product (AGEs) levels were found to be increased in the serum of young women with PCOS [68]. In addition, increased immunostaining of AGEs and their receptors (RAGE) was observed in the different compartments of the ovarian tissue in polycystic ovaries [69]. Specifically, serum concentrations of some AGEs and intraovarian deposition of AGEs could potentially contribute to alteration in oocyte function, fertilization, and embryo development [70]. Another commonly cited factor, seen in unexplained reproductive abnormalities, is chronic stress. As already mentioned, chronic stress could induce hypothalamic inflammation in mice [71]. Even in pregnancy, chronic stress can lead to elevated concentrations of cortisol and insulin [72]. It has been suspected that chronic stress, via hypothalamic inflammation, could potentially activate the hypothalamus–pituitary axis and result in reproductive, metabolic, and endocrine dysregulation.
In this review, data are presented showing that HFD can induce hypothalamic inflammation. In addition, many studies have shown that an HFHS diet is associated with menstrual disorders, anovulation, and difficulties in fertilization in women diagnosed with PCOS. Thus, it appears that diet-induced hypothalamic inflammation may contribute to the endocrine pathogenesis of PCOS. Of note, the extent and pattern of hypothalamic inflammation in these patients is not yet fully studied. The preoptic area as well as the arcuate nucleus, which modulate secretion of GnRH, could be a potential target of hypothalamic inflammation.
3.3. Potential Mechanisms of Hypothalamic Inflammation-Associated Psychiatric Disorders
As stated before, PCOS is associated with depression and anxiety disorders [59]. Chronic exposure to intense stress is associated with an elevation of inflammatory molecules in the hypothalamus, altering the activity of the HPA axis, ultimately leading to glucocorticoid resistance [73][74]. The latter might participate in the pathogenesis of psychiatric disorders, including depression [75][76]. Long chronic stress induces glucocorticoid resistance in CRH neurons in PVN, thus disrupting the negative feedback and altering the physiological function of the HPA axis [75]. Along with this, repeated administration of recombined IL-6 in humans leads to a blunted ACTH response [77]. This resetting of the HPA axis has also been observed in melancholic depression. Glucocorticoid resistance decreases the availability of neuroprotective factors and increases the expression of inflammatory molecules in the hypothalamus [74]. Moreover, increased glucocorticoid concentrations induce neurotoxicity and subsequent atrophy in the hippocampus, which is crucially involved in the pathogenesis of depressive disorders [78]. Overall, these changes might lead to alterations in neurotransmission, neurodegeneration, nerve cell death, and ultimately depression [79]. Indeed, recent studies indicate that hypothalamic inflammation might also be associated with stress exposure and psychiatric diseases, including depressive disorder [80].
It seems that hypothalamic inflammation could be associated with the psychiatric profile described in women with PCOS. The exact pathophysiology of hypothalamic inflammation-induced psychiatric disorders in PCOS women needs to be further elucidated.
References
- Markakis, E.A. Development of the neuroendocrine hypothalamus. Front. Neuroendocrinol. 2002, 23, 257–291.
- Lechan, R.M.; Toni, R. Functional Anatomy of the Hypothalamus and Pituitary; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; Available online: https://www.ncbi.nlm.nih.gov/books/NBK279126/ (accessed on 17 December 2020).
- Marshall, J.C.; Dalkin, A.C.; Haisenleder, D.J.; Griffin, M.L.; Kelch, R.P. GnRH pulses—The regulators of human reproduction. Trans. Am. Clin. Climatol. Assoc. 1992, 104, 31–46.
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867.
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.A.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199.
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127.
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162.
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Näslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nat. Cell Biol. 2008, 453, 783–787.
- Boden, G. Obesity and Free Fatty Acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646.
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased Number of Islet-Associated Macrophages in Type 2 Diabetes. Diabetes 2007, 56, 2356–2370.
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated Fatty Acid and TLR Signaling Link β Cell Dysfunction and Islet Inflammation. Cell Metab. 2012, 15, 518–533.
- Escobar-Morreale, H.F. Polycystic ovary syndrome: Definition, aetiology, diagnosis and treatment. Nat. Rev. Endocrinol. 2018, 14, 270–284.
- Chang, J.; Azziz, R.; Legro, R.; Dewailly, D.; Franks, S.; Tarlatzis, R.; Fauser, B.; Balen, A.; Bouchard, P.; Dalgien, E.; et al. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil. Steril. 2004, 81, 19–25.
- Peigné, M.; Dewailly, D. Long term complications of polycystic ovary syndrome (PCOS). Ann. d’Endocrinol. 2014, 75, 194–199.
- Sanchez-Garrido, M.A.; Tena-Sempere, M. Metabolic dysfunction in polycystic ovary syndrome: Pathogenic role of androgen excess and potential therapeutic strategies. Mol. Metab. 2020, 35, 100937.
- Goodarzi, M.O.; Dumesic, D.A.; Chazenbalk, G.; Azziz, R. Polycystic ovary syndrome: Etiology, pathogenesis and diagnosis. Nat. Rev. Endocrinol. 2011, 7, 219–231.
- Antoniou-Tsigkos, A.; Macut, D.; Mastorakos, G. Physiopathology, Diagnosis, and Treatment of Secondary Female Hypogonadism BT-Hypothalamic-Pituitary Diseases; Casanueva, F.F., Ghigo, E., Eds.; Springer International Publishing: Cham, Switzerland; Berlin/Heidelberg, Germany, 2018; pp. 247–287. ISBN 978-3-319-44444-4.
- Valsamakis, G.; Lois, K.; Kumar, S.; Mastorakos, G. Metabolic and other effects of pioglitazone as an add-on therapy to metformin in the treatment of polycystic ovary syndrome (PCOS). Hormones 2013, 12, 363–378.
- Yildiz, B.O.; Bozdag, G.; Yapici, Z.; Esinler, I.; Yarali, H. Prevalence, phenotype and cardiometabolic risk of polycystic ovary syndrome under different diagnostic criteria. Hum. Reprod. 2012, 27, 3067–3073.
- Chaudhari, A.P.; Mazumdar, K.; Mehta, P.D. Anxiety, Depression, and Quality of Life in Women with Polycystic Ovarian Syndrome. Indian J. Psychol. Med. 2018, 40, 239–246.
- Lee, I.; Cooney, L.G.; Saini, S.; Sammel, M.D.; Allison, K.C.; Dokras, A. Increased odds of disordered eating in polycystic ovary syndrome: A systematic review and meta-analysis. Eat. Weight. Disord. 2018, 24, 787–797.
- Laven, J.S.E.; Imani, B.; Eijkemans, M.J.C.; Fauser, B.C.J.M. New Approach to Polycystic Ovary Syndrome and Other Forms of Anovulatory Infertility. Obstet. Gynecol. Surv. 2002, 57, 755–767.
- Kyrkou, G.; Trakakis, E.; Attilakos, A.; Panagopoulos, P.; Chrelias, C.; Papadimitriou, A.; Vaggopoulos, V.; Alexiou, E.; Mastorakos, G.; Lykeridou, A.; et al. Metabolic syndrome in Greek women with polycystic ovary syndrome: Prevalence, characteristics and associations with body mass index. A prospective controlled study. Arch. Gynecol. Obstet. 2015, 293, 915–923.
- Kim, J.J.; Choi, Y.M. Dyslipidemia in women with polycystic ovary syndrome. Obstet. Gynecol. Sci. 2013, 56, 137–142.
- Palomba, S.; Falbo, A.; Chiossi, G.; Muscogiuri, G.; Fornaciari, E.; Orio, F.; Tolino, A.; Colao, A.; La Sala, G.B.; Zullo, F. Lipid profile in nonobese pregnant women with polycystic ovary syndrome: A prospective controlled clinical study. Steroids 2014, 88, 36–43.
- Blank, S.; McCartney, C.; Marshall, J. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum. Reprod. Updat. 2006, 12, 351–361.
- Yao, B.; Liu, H.-Y.; Gu, Y.-C.; Shi, S.-S.; Tao, X.-Q.; Li, X.-J.; Ge, Y.-F.; Cui, Y.-X.; Yang, G.-B. Gonadotropin-releasing hormone positively regulates steroidogenesis via extracellular signal-regulated kinase in rat Leydig cells. Asian J. Androl. 2011, 13, 438–445.
- Han, S.-K.; Gottsch, M.L.; Lee, K.J.; Popa, S.M.; Smith, J.T.; Jakawich, S.K.; Clifton, D.K.; Steiner, R.A.; Herbison, A.E. Activation of Gonadotropin-Releasing Hormone Neurons by Kisspeptin as a Neuroendocrine Switch for the Onset of Puberty. J. Neurosci. 2005, 25, 11349–11356.
- Marshall, J.C.; Dunaif, A. Should all women with PCOS be treated for insulin resistance? Fertil. Steril. 2012, 97, 18–22.
- Tosi, F.; Negri, C.; Perrone, F.; Dorizzi, R.; Castello, R.; Bonora, E.; Moghetti, P. Hyperinsulinemia Amplifies GnRH Agonist Stimulated Ovarian Steroid Secretion in Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 1712–1719.
- Baillargeon, J.P.; Nestler, J.E. Commentary: Polycystic ovary syndrome: A syndrome of ovarian hypersensitivity to insulin? J. Clin. Endocrinol. Metab. 2006, 91, 22–24.
- Schneider, J.E. Energy balance and reproduction. Physiol. Behav. 2004, 81, 289–317.
- Chou, S.H.; Mantzoros, C.S. 20 YEARS OF LEPTIN: Role of leptin in human reproductive disorders. J. Endocrinol. 2014, 223, T49–T62.
- Gordon, C.M.; Ackerman, K.E.; Berga, S.L.; Kaplan, J.R.; Mastorakos, G.; Misra, M.; Murad, M.H.; Santoro, N.F.; Warren, M.P. Functional Hypothalamic Amenorrhea: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2017, 102, 1413–1439.
- Quennell, J.H.; Howell, C.S.; Roa, J.; Augustine, R.A.; Grattan, D.R.; Anderson, G.M. Leptin Deficiency and Diet-Induced Obesity Reduce Hypothalamic Kisspeptin Expression in Mice. Endocrinology 2011, 152, 1541–1550.
- Brown, R.; Imran, S.; Ur, E.; Wilkinson, M. KiSS-1 mRNA in adipose tissue is regulated by sex hormones and food intake. Mol. Cell. Endocrinol. 2008, 281, 64–72.
- Araujo, E.P.; Moraes, J.C.; Cintra, D.E.; Velloso, L.A. Mechanisms in endocrinology: Hypothalamic inflammation and nutrition. Eur. J. Endocrinol. 2016, 175, R97–R105.
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic Inflammation in the Control of Metabolic Function. Annu. Rev. Physiol. 2015, 77, 131–160.
- Gropp, E.; Shanabrough, M.; Borok, E.; Xu, A.W.; Janoschek, R.; Buch, T.; Plum, L.; Balthasar, N.; Hampel, B.; Waisman, A.; et al. Agouti-related peptide–expressing neurons are mandatory for feeding. Nat. Neurosci. 2005, 8, 1289–1291.
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of Melanocortin Pathways in the Control of Food Intake and Energy Expenditure. Cell 2005, 123, 493–505.
- Mastorakos, P.; McGAVERN, D.B. The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol. 2019, 4, eaav0492.
- Papargyri, P.; Zapanti, E.; Salakos, N.; Papargyris, L.; Bargiota, A.; Mastorakos, G. Links between HPA axis and adipokines: Clinical implications in paradigms of stress-related disorders. Expert Rev. Endocrinol. Metab. 2018, 13, 317–332.
- Kleinridders, A.; Könner, A.C.; Bruning, J.C. CNS-targets in control of energy and glucose homeostasis. Curr. Opin. Pharmacol. 2009, 9, 794–804.
- Könner, A.C.; Klöckener, T.; Brüning, J.C. Control of energy homeostasis by insulin and leptin: Targeting the arcuate nucleus and beyond. Physiol. Behav. 2009, 97, 632–638.
- Williams, K.W.; Margatho, L.O.; Lee, C.E.; Choi, M.; Lee, S.; Scott, M.M.; Elias, C.F.; Elmquist, J.K. Segregation of Acute Leptin and Insulin Effects in Distinct Populations of Arcuate Proopiomelanocortin Neurons. J. Neurosci. 2010, 30, 2472–2479.
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.-K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of Central Melanocortin Receptors in Vitro and in Vivo by Agouti-Related Protein. Science 1997, 278, 135–138.
- André, C.; Guzman-Quevedo, O.; Rey, C.; Rémus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2016, 66, 908–919.
- Dalvi, P.S.; A Chalmers, J.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158.
- Tsaousidou, E.; Paeger, L.; Belgardt, B.F.; Pal, M.; Wunderlich, C.M.; Brönneke, H.; Collienne, U.; Hampel, B.; Wunderlich, F.T.; Schmidt-Supprian, M.; et al. Distinct Roles for JNK and IKK Activation in Agouti-Related Peptide Neurons in the Development of Obesity and Insulin Resistance. Cell Rep. 2014, 9, 1495–1506.
- Ernst, M.B.; Wunderlich, C.M.; Hess, S.; Paehler, M.; Mesaros, A.; Koralov, S.B.; Kleinridders, A.; Husch, A.; Münzberg, H.; Hampel, B.; et al. Enhanced Stat3 Activation in POMC Neurons Provokes Negative Feedback Inhibition of Leptin and InsulinSignaling in Obesity. J. Neurosci. 2009, 29, 11582–11593.
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569.
- Yang, L.; Qi, Y.; Yang, Y. Astrocytes Control Food Intake by Inhibiting AGRP Neuron Activity via Adenosine A1 Receptors. Cell Rep. 2015, 11, 798–807.
- Yan, J.; Zhang, H.; Yin, Y.; Li, J.; Tang, Y.; Purkayastha, S.; Li, L.; Cai, D. Obesity- and aging-induced excess of central transforming growth factor-β potentiates diabetic development via an RNA stress response. Nat. Med. 2014, 20, 1001–1008.
- De Araujo, E.P.; De Souza, C.T.; Velloso, L. Atypical transforming growth factor–β signaling in the hypothalamus is linked to diabetes. Nat. Med. 2014, 20, 985–987.
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in Hypothalamic AgRP Neurons Regulates Food Intake and Energy Balance. Cell Metab. 2011, 14, 173–183.
- Kaushik, S.; Arias, E.; Kwon, H.; Lopez, N.M.; Athonvarangkul, D.; Sahu, S.; Schwartz, G.J.; E Pessin, J.; Singh, R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012, 13, 258–265.
- Schneeberger, M.; Altirriba, J.; García, A.; Esteban, Y.; Castaño, C.; García-Lavandeira, M.; Alvarez, C.V.; Gomis, R.; Claret, M. Deletion of miRNA processing enzyme Dicer in POMC-expressing cells leads to pituitary dysfunction, neurodegeneration and development of obesity. Mol. Metab. 2013, 2, 74–85.
- Meng, Q.; Cai, D. Defective Hypothalamic Autophagy Directs the Central Pathogenesis of Obesity via the IκB Kinase β (IKKβ)/NF-κB Pathway. J. Biol. Chem. 2011, 286, 32324–32332.
- Portovedo, M.; Ignacio-Souza, L.M.; Bombassaro, B.; Coope, A.; Reginato, A.; Razolli, D.S.; Torsoni, M.A.; Torsoni, A.S.; Leal, R.F.; Velloso, L.A.; et al. Saturated Fatty Acids Modulate Autophagy’s Proteins in the Hypothalamus. PLoS ONE 2015, 10, e0119850.
- Mendes, N.F.; Kim, Y.-B.; Velloso, L.A.; De Araujo, E.P. Hypothalamic Microglial Activation in Obesity: A Mini-Review. Front. Neurosci. 2018, 12, 846.
- Han, C.; Rice, M.W.; Cai, D. Neuroinflammatory and autonomic mechanisms in diabetes and hypertension. Am. J. Physiol. Metab. 2016, 311, E32–E41.
- Ulrich-Lai, Y.M.; Herman, J.P. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 2009, 10, 397–409.
- Scherer, T.; Lindtner, C.; Zielinski, E.; O’Hare, J.; Filatova, N.; Buettner, C. Short Term Voluntary Overfeeding Disrupts Brain Insulin Control of Adipose Tissue Lipolysis. J. Biol. Chem. 2012, 287, 33061–33069.
- Van Santbrink, E.J.P.; Eijkemans, M.J.; Laven, J.S.E.; Fauser, B.C.J.M. Patient-tailored conventional ovulation induction algorithms in anovulatory infertility. Trends Endocrinol. Metab. 2005, 16, 381–389.
- Rivest, S.; Lee, S.; Attardi, B.; Rivier, C. The chronic intracerebroventricular infusion of interleukin-1 beta alters the activity of the hypothalamic-pituitary-gonadal axis of cycling rats. I. Effect on LHRH and gonadotropin biosynthesis and secretion. Endocrinology 1993, 133, 2424–2430.
- Hohos, N.M.; Skaznik-Wikiel, M.E. High-Fat Diet and Female Fertility. Endocrinology 2017, 158, 2407–2419.
- Roberts, J.S.; Perets, R.A.; Sarfert, K.S.; Bowman, J.J.; Ozark, P.A.; Whitworth, G.B.; Blythe, S.N.; Toporikova, N. High-fat high-sugar diet induces polycystic ovary syndrome in a rodent model†. Biol. Reprod. 2017, 96, 551–562.
- Diamanti-Kandarakis, E.; Katsikis, I.; Piperi, C.; Kandaraki, E.; Piouka, A.; Papavassiliou, A.G.; Panidis, D. Increased serum advanced glycation end-products is a distinct finding in lean women with polycystic ovary syndrome (PCOS). Clin. Endocrinol. 2008, 69, 634–641.
- DiamantiKandarakis, E.; Piperi, C.; Patsouris, E.; Korkolopoulou, P.; Panidis, D.; Pawelczyk, L.; Papavassiliou, A.G.; Duleba, A.J. Immunohistochemical localization of advanced glycation end-products (AGEs) and their receptor (RAGE) in polycystic and normal ovaries. Histochem. Cell Biol. 2007, 127, 581–589.
- Tatone, C.; Eichenlaub-Ritter, U.; Amicarelli, F. Dicarbonyl stress and glyoxalases in ovarian function. Biochem. Soc. Trans. 2014, 42, 433–438.
- Wohleb, E.S.; Fenn, A.M.; Pacenta, A.M.; Powell, N.D.; Sheridan, J.F.; Godbout, J.P. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 2012, 37, 1491–1505.
- Valsamakis, G.; Chrousos, G.; Mastorakos, G. Stress, female reproduction and pregnancy. Psychoneuroendocrinology 2019, 100, 48–57.
- McEwen, B.S. Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain. Physiol. Rev. 2007, 87, 873–904.
- Merkulov, V.M.; Merkulova, T.I.; Bondar, N.P. Mechanisms of brain glucocorticoid resistance in stress-induced psychopathologies. Biochemistry 2017, 82, 351–365.
- Duque, E.D.A.; Munhoz, C.D. The Pro-inflammatory Effects of Glucocorticoids in the Brain. Front. Endocrinol. 2016, 7, 78.
- Burfeind, K.G.; Michaelis, K.A.; Marks, D.L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 2016, 54, 42–52.
- Mastorakos, G.; Chrousos, G.P.; Weber, J.S. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J. Clin. Endocrinol. Metab. 1993, 77, 1690–1694.
- Tatomir, A.; Micu, C.; Crivii, C.B. THE IMPACT OF STRESS AND GLUCOCORTICOIDS ON MEMORY. Clujul Med. 2014, 87, 3–6.
- Cernackova, A.; Durackova, Z.; Trebaticka, J.; Mravec, B. Neuroinflammation and depressive disorder: The role of the hypothalamus. J. Clin. Neurosci. 2020, 75, 5–10.
- Tafet, G.E.; Nemeroff, C.B. The Links Between Stress and Depression: Psychoneuroendocrinological, Genetic, and Environmental Interactions. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 77–88.


