 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Enrique Madruga | + 5104 word(s) | 5104 | 2021-02-04 03:48:50 | | | |
| 2 | Ana Martinez | Meta information modification | 5104 | 2021-02-08 12:53:48 | | | | |
| 3 | Camila Xu | Meta information modification | 5104 | 2021-02-10 09:25:00 | | |
Video Upload Options
Mitophagy, as a selective variant of autophagy, is characterized by molecular mechanisms that allow selective degradation of mitochondria.
1. Mitophagy Machinery: A Mitochondrial-Selective Autophagy
First, autophagy must be regarded as a highly dynamic process along time and space, where a great diversity of proteins is involved to ensure its regulation. Two classical autophagy master keys are the mammalian targets of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK). The first one is in charge of the inhibition of this pathway through the phosphorylation of Unc-51 like autophagy activating kinase 1 (ULK1) when autophagy is not needed (e.g., in abundant nutrient-rich situations). Alternatively, AMPK also phosphorylates ULK1 resulting in its activation and triggering autophagy. Thus, a balance in both kinase activities is needed to achieve correct levels of this catabolic pathway [1]. Autophagy begins with the formation of the phagophore, a lipid membrane that selectively elongates over the cargo that must be degraded, whether mitochondrion or another kind of target [2]. In any case, cargo is marked with microtubule-associated proteins 1A/1B light chain 3B (LC3) in order to discriminate those to be degraded. After that, the phagophore becomes a double-membrane vesicle called an autophagosome, probably through the fission of the pre-autophagosomal membrane [3]. The double-membrane vesicle is transported along the axon to the soma, where huge populations of lysosomes are found. Finally, the autophagosome merges with the lysosome, generating the autolysosome, where the cargo is degraded by hydrolytic enzymes [4].
Mitophagy, as a selective variant of autophagy, is characterized by molecular mechanisms that allow selective degradation of mitochondria. The most studied mechanism is the PINK1/PARKIN pathway. PTEN-induced kinase 1 (PINK1) is a cytoplasmatic kinase able to phosphorylate different substrates, including ubiquitin and PARKIN. On the other hand, PARKIN is a E3 ubiquitin-ligase [5][6]. In the case of a healthy mitochondrion, PINK1 is imported into the mitochondrion through the translocases of the outer and inner membrane (TOM and TIM, respectively) complexes. Afterwards, PINK1 is degraded inside the organelle [7]. However, when the mitochondrion is damaged, TOM and TIM complexes are not able to import PINK1, which is the kinase accumulated on the mitochondrial surface. PINK1 phosphorylates ubiquitin, turns it into an allosteric modulator of PARKIN and prompts its recruitment [8]. PINK1 is also able to phosphorylate outer membrane proteins such as mitofusin 2 (MFN2), facilitating the recruitment of the ubiquitin-ligase as well [9]. Subsequently, PINK1 phosphorylates PARKIN, producing a conformational change and discovering its ubiquitin-ligase activity on various proteins such as MFN1, MFN2, voltage-dependent anion channel 1 (VDAC1), and MIRO1 [7][8]. Therefore, ubiquitin is both a substrate and a PARKIN modulator. Once the mitochondrion is polyubiquitinated, autophagy receptors such us optineurin (OPTN), p62, nuclear dot protein 52 (NDP52), TAX1 binding protein 1 (TAX1BP1), and neighbor of BRCA1 gene 1 (NBR1) facilitate the formation of autophagosome around the defective mitochondrion [10][11]. These receptors have at least two domains: one responsible for recognizing ubiquitin and another able to link LC3 (LC3-interaction region, LIR motif). This way, only ubiquitinated mitochondria are marked LC3-positive and, hence, are degraded (Figure 2).
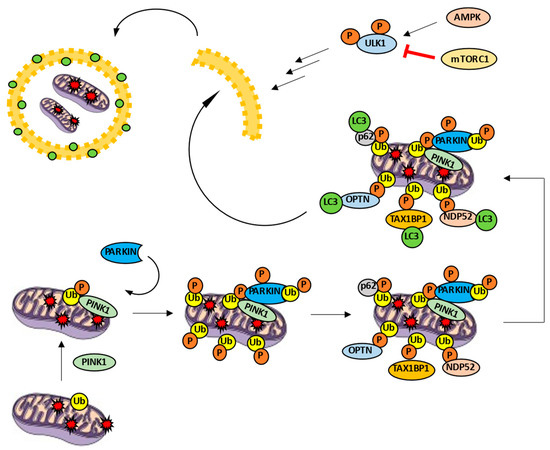
Figure 2. PINK1/PARKIN pathway in mitophagy activity. PINK1 is accumulated in the mitochondrial surface and phosphorylates ubiquitin, recruiting PARKIN ubiquitin ligase. PARKIN is phosphorylated and increases the number of ubiquitins. Autophagy receptors such as OPTN, p62, NDP52 or TAX1BP1 link to the phospho-ubiquitin and LC3, promoting the formation of the autophagosome around the cargo.
Interestingly, PINK1 is able to recruit OPTN and NDP52 by means of phospho-ubiquitin without PARKIN activity [12]. Moreover, knockout models point to only OPTN and NDP52 as strictly necessary autophagy receptors to trigger mitophagy through PINK/PARKIN pathway and overshadow the rest of receptors [12]. Besides, OPTN and NDP52 receptors are able to recruit ULK1 by themselves and promote phagophore formation [12]. It is worth nothing that the role of p62 as a possible dispensable receptor keeps generating controversy [11]. All the above leads us to think that PARKIN functions as a PINK1 modulator: PARKIN facilitates an increase of ubiquitins on the outer mitochondrial membrane. Then, these ubiquitins can be phosphorylated by PINK1 in order to recruit a greater number of autophagy receptors. Without this positive feedback mechanism, mitophagy levels would never be enough due to the low constitutive amount of ubiquitin in the mitochondrion. This explains, at least in part, the mild phenotype observed in PARKIN−/− models [13].
The paragraphs above describe the PINK1/PARKIN-dependent mitophagy. However, other pathways to trigger mitophagy have been reported in the last years. First of all, LC3 is considered a canonical ATG8 yeast homologue in humans and is extremely useful in autophagy studies. However, other ATG8 homologues have been reported in mammals with implications for autophagy. Indeed, the ATG8 homologue gamma-aminobutyric acid receptor-associated protein-like 1 (GABARAPL1) is highly expressed in motoneurons [14]. On the other hand, cardiolipin can interact with LC3 without PINK1/PARKIN activity. Under mitochondrial damage conditions, inner mitochondrial membrane lipid cardiolipin can flow to the outer membrane and initiate mitophagy [15]. Moreover, several proteins on the outer mitochondrial membrane contain LIR motifs, which ensure a direct interaction between defective mitochondria and LC3. Some examples are autophagy and BECLIN 1 regulator 1 (AMBRA1) and FUN14 domain containing protein 1 (FUNDC1) [7][15][16]. Finally, there are other ubiquitin-ligase proteins like PARKIN that are capable of participating in mitophagy such as the mitochondrial ubiquitin-ligase activator of NF-kB (MULAN) and the membrane-associated ring finger 5 ubiquitin-ligase (MARCH5) [15]. The fact that PINK1/PARKIN-dependent pathway is not responsible for all mitophagy activity brings new pieces to the unsolved puzzle of mitochondrial quality control. Future investigations may declare these alternative pathways as interesting targets in mitophagy-altered diseases.
2. Mitophagy and ALS
As we exposed before, mitochondrial quality control is tightly regulated through the orchestrated action of several pathways. Taking into consideration that maintaining a healthy pool of mitochondria in neurons is something critical in order to ensure their functionality, mitophagy mechanisms must work properly to guarantee a reduced level of defective mitochondria. Nevertheless, the large number of proteins involved in mitophagy provides several hot points where diseases can be triggered. In the case of ALS, many defects in mitochondria degradation have been documented in the last years. The role of PINK1/PARKIN has been extensively studied in multiple neurodegenerative diseases and there is a certain consensus about its neuroprotective action. However, its role in ALS still produces controversy. TDP-43 is responsible, under physiological conditions, for regulating the transcription of many mRNAs, including PARKIN mRNA. The spinal cords of sALS patients with TDP-43 accumulation has been found to show a decrease in PARKIN levels [17]. SOD1G93A mice models exhibit a highly stimulated mitophagy at early stages of the disease despite a reduction in spinal cord PARKIN levels [18]. When PARKIN is knocked out in this model, mice show an increase in survival rate. The authors argue that PARKIN could have a short-term neuroprotective effect, but a long continued mitophagy activity would lead to defective mitochondrial machinery and be detrimental in the long term. In flies expressing both FUS and FUSP525L, PINK1 and PARKIN levels are increased, while their reduction by siRNA improves neurodegenerative phenotypes caused by FUS [19]. Nonetheless, a recent study on flies shows the reversal of locomotive defects caused by FUS through PARKIN expression [20].
Autophagy receptor defects have been found in several ALS types. There are about forty different ALS-related OPTN mutations, which contribute to 1% of fALS cases, although they can also be observed in sALS patients [21]. For example, ALS-related mutations such as E478G and Q398X prevent OPTN-ubiquitin binding and impair mitophagy [10][22]. Other functions have been associated with this receptor; since OPTN is able to bind LC3 and myosin VI, it is able to regulate autophagosome flux through the neuron [23]. Moreover, E478G and Q398W mutations inhibit the fusion of autophagosomes with lysosomes in NSC-34 cells, a widely used motoneuron model [10]. Recently, an interesting link between SOD1 aggregates and OPTN was also reported: SOD1 aggregates retain OPTN and interfere with the mitophagy machinery [24]. On the other hand, mutations in SQSTM1 (the gene that encodes for p62) embrace 1% of fALS cases [21]. Its role in protein degradation is widely known and, in the same way as LC3, p62 can be used as a marker in autophagy defect studies. In addition, p62 can be found in protein aggregates of some ALS patients and its depletion has been shown to shorten life expectancy in SOD1H46R and SOD1G93A transgenic models [25]. In the last years, the relevance of the TANK-binding kinase 1 (TBK1) as a mitophagy modulator has been highlighted. This serine/threonine kinase is able to phosphorylate p62, NDP52, and OPTN [11], increasing its ability to link ubiquitin and LC3. Autophagic receptors are capable of binding ubiquitin, resulting in low mitophagy activity. This binding causes TBK1 activation and the consequent phosphorylation of autophagic receptors, increasing their activity [26]. Besides, TBK1 induces TBK1-OPTN retention on the surface of damaged mitochondria. More than eighty ALS-related TBK1 mutations have been reported [11]. For example, p.690-713 deletion of TBK1 at C-terminal impairs its binding with OPTN and compromises mitophagy [27]. TBK1 may also play a regulatory role in the onset of mitophagy since C9orf78 is able to promote mitophagy through the interaction with ULK1 complex due to its binding with the Smith–Magenis syndrome chromosome region candidate 8 (SMCR8), a guanine nucleotide exchange protein. C9orf78 is activated by TBK1 through an SMCR8 TBK1-dependent phosphorylation [28]. TBK1 may also alter later processes of mitophagy such as the transport of autophagosomes. This can be explained due to the role of TBK1 in the regulation of microtubule polarization during mitosis, as well as the regulation of cytoplasmatic levels of dynein [11].
As we exposed before, one of the strongest indicators correlating mitophagy and ALS is the fact that several ALS-related mutations lead to the misfunction of proteins such as OPTN, p62, or TBK1 and, therefore, an inefficient mitophagy. However, the majority of sALS patients do not present these kinds of mutations, raising the following question: is mitophagy modulation an interesting topic for all the ALS patients or only for those who present mitophagy mutations? Mitochondria can be considered as a convergence point in ALS pathology. Mitochondria defects can be associated with most of the pathways involved in ALS, as the accumulation of dysfunctional mitochondria is related to inadequate ATP generation, high ROS levels, altered Ca2+ buffering, and cell death. Thus, ensuring a healthy pool of mitochondria is crucial to improve ALS pathology, which includes correct mitophagy levels, regardless of whether patients present mitophagy mutations or not. On another note, TDP-43 has been reported to alter several mitochondrial pathways such as mitochondrial dynamics, mitochondrial trafficking, and energetic metabolism [29]. Recently, it has also been documented that TDP-43M337V mice show increased levels of TDP-43 in mitochondria with corresponding mitochondrial damage and cell death. Surprisingly, blocking the link between TDP-43 and mitochondrion is enough to alleviate the neuronal loss and mitochondrial dysfunction [30]. Thus, preventing TDP-43-mitochondrion interaction and degrading these defective mitochondria may be a promising approach in order to treat ALS. Since TDP-43 aggregates are found in the vast majority of patients, ensuring an adequate mitophagy activity in order to maintain a healthy pool of mitochondria is a promising opportunity for ALS treatment.
2.1. Mitophagy and Neuroinflammation in ALS
Mitophagy and neuroinflammation are interestingly overlapped because of the pleiotropic character of the genes involved. Neuroinflammation is usually related to the reaction of glial cells (astrocytes, microglia, and oligodendrocytes) and circulating immune cells (such as lymphocytes) to infection, injury, or degeneration. Therefore, long-term overproduction of inflammatory mediators eventually produces cell death. A wide consensus has been achieved on the correlation between neuroinflammation and neurodegenerative diseases. The relation between the OPTN autophagy receptor and neuroinflammation is increasingly striking. Knockdown [31] and knockout [32] models show an OPTN inhibitory role on nuclear factor-kappa B (NF-kB) in vitro. Interestingly, ALS-related mutations Q398X and E378G do not exhibit this kind of activity [10]. On the other hand, TBK1 is known to regulate neuroinflammation through type I interferon (IFN). This kinase acts downstream of toll-like receptor 3 (TLR3), which is implicated in the immune response, as TBK1 is an activator of interferon regulatory factor 3 (IRF3) and results in an increase of IFNα, β, and γ [33]. Since the treatment of ALS patients with IFNα and β leads to strong inflammation [34][35], TBK1 activity is expected to increase neuroinflammation and have a negative role in ALS. In contrast, TBK1 could have a protective role in these glial cells. Circulating T-cells are thought to stabilize microglia and reduce proinflammatory cytokines levels. TBK1-/- T cells show a reduced migration to the central nervous system (CNS), which could reduce the number of T cells infiltrated in the CNS and result in increased damage from ALS [36]. Besides, it has been documented that TBK1 is a natural endogenous inhibitor of receptor-like protein kinase 1 (RLPK1), which is highly related to apoptosis and inflammation events [37]. The pleiotropic nature of TBK1 should be carefully considered before declaring any statement. To sum up, since many alterations related to neurodegeneration can lead to mitophagy defects and neuroinflammation, the overlapping field between both rises as an interesting topic of research.
2.2. Mitophagy as Target
In the recent years, a growing number of articles have been reported to link ALS, autophagy, and mitophagy. An increase in autophagosomes and autolysosomes highly rich in mitochondria in the spinal cords of ALS patients have been described [38]. The ablation of genes involved in autophagy is enough to trigger neurodegeneration in murine models [39][40] and, as it is described above, multiple ALS-related genes are directly involved in mitophagy pathways such as OPTN, TBK1, or SQSTM1. Therefore, many autophagy-stimulating drugs have been probed in ALS models and studied to determine their pharmacological action. Several molecules are able to reduce TDP-43 aggregates by means of autophagy stimulation, some examples are rapamycin or trehalose. However, the effects of autophagy modulators rely heavily upon the ALS model used [41]. Verapamil has been shown to delay the onset of the disease, increase life expectancy, regain the functionality of affected motor neurons, and reduce the aggregation of SOD1 by increasing autophagy in the SOD1G93A model [42]. At the same time, its ability to enhance autophagosome–lysosome fusion is well known [43]. Long-term rilmenidine use, a common anti-hypertensive, promotes mTOR-independent autophagy in the spinal cord of SOD1G93A, hardly reducing the onset of hind-limb paralysis and producing depletion of mitochondrial levels; this is accompanied by an accelerated motoneuron degeneration [44]. Autophagic stimulation of clemastine shows beneficial effects only in the short term, and presents no effect on survival or disease progression in the long term [45]. Rapamycin shows null or negative effects on SOD1 models, but beneficial effects in a TDP-43 model [41]. This issue is more raveled with respect to trehalose, where contradictory data can be found. In 2013, trehalose was reported to show beneficial effects in the disease progression of a SOD1G86R mouse model [46], in the same way that SOD1G93A mouse was reported [47]. Two years later, by contrast, trehalose was reported to perform beneficial roles only in early stages in the SOD1G93A model [48]. Taking all these data into consideration, the huge number of ALS models, and the diversity at the time of administrating the drug, making absolute statements about this issue is risky. The paragraphs below try to bring light to the role of mitochondria in this brainteaser.
2.2.1. Mitophagy Is a Highly (Temporal) Dynamic Puzzle
Mitophagy is a fundamental mechanism in the maintenance of homeostasis, so the elimination of defective mitochondria is critical for the proper functioning of the neuron. However, an excess of mitophagy activity can result in the reduction of functional mitochondria, so this strategy could not only be inefficient but even harmful. Mitophagy activity must be in balance with the mitochondrial biogenesis or mitochondriogenesis, a set of mechanisms aimed at increasing mitochondrial machinery. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is one of the major transcription factors that regulate mitochondriogenesis. Although low levels of PGC-1α have been detected in animal models and ALS patients [49], mRNA PGC1-α was progressively increased until 60 days of life in SOD1G93A mouse and decreased from 90 days until death [18]. Keeping this in mind, one possible hypothesis that may explain this is a progressive imbalance between mitophagy and mitochondriogenesis. At early stages of the disease, mitophagy induction may prove beneficial as mitochondriogenesis provides new and healthy mitochondria, maintaining appropriate levels of functional mitochondria. In contrast, mitophagy induction at late stages would result in a deficit of mitochondrial machinery due to an inefficient biogenesis. On the other hand, the SOD1G93A transgenic model shows a markedly progressive deficiency of the endolysosomal system activity, that is, the set of mechanisms aimed to transport autophagosomes and merge them with the lysosomes [50]. Time-lapse studies have shown that the temporal dynamics of neuronal mitophagy are very slow, in which lysosomal fusion and acidification are the rate-limiting stages [51]. Indeed, several ALS-related mutations in genes such as C9orf72, ALS2, or CHMPB2 are involved in autophagosome maturation defects and their fusion with the lysosomes [41]. All of this defines a hypothetical model where mitophagy induction may be detrimental at late stages of neurodegeneration due to an accumulation of nonfunctional vacuolated organelles and the inability of the neuron to provide new healthy mitochondria (Figure 3). According to the information exposed before, enhancing lysosomal function could be an interesting strategy over forcing general autophagy. This shows the highly dynamic nature of this kind of pathway and the need to display not only a strong autophagy or mitophagy, but a time-coordinated mitochondrial quality control. It is therefore not surprising that autophagy inductors, such as lithium [52] or resveratrol [53], also increase mitochondrial biogenesis. It is of note that a sole increase in mitochondriogenesis rate without autophagy stimulation cannot extend survival in ALS mouse models despite showing improvements in muscle activity [54]. Ultimately, better knowledge about autophagy machinery based on disease stage would provide clues about which part of mitochondrial quality control we need to enhance.
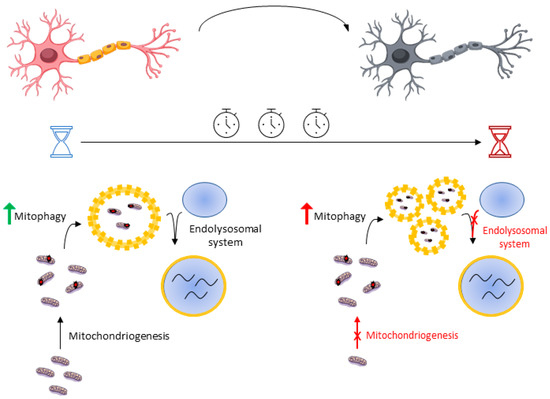
Figure 3. Mitophagy induction shows different effects at early and late stages. Boosting mitophagy may be beneficial in the short term as mitochondriogenesis and the endolysosomal system work properly. However, this strategy may be detrimental in the long term due to progressive defects in both mitochondriogenesis and the endolysosomal system, resulting in the accumulation of nonfunctional vacuolated organelles.
2.2.2. Mitophagy Is a Highly (Neuron-Type) Dynamic Puzzle
In 2006 and in parallel, CNS-selective deletion of autophagy models were generated in order to study the role of autophagy in neurodegenerative diseases. As is well known, deletion of ATG5 [39] or ATG7 [40] (both essential genes for autophagy activity) in the CNS leads to neurodegenerative phenotypes, so the spotlight was put on autophagy machinery in the race to discover an effective treatment for ALS. Although ALS was initially considered as a pure neurodegenerative disease, subsequent reports attributed a muscle-disease nature, since motoneuron degeneration in ALS models is preceded by denervation of the neuromuscular junction (NMJ) [55][56]. Therefore, the NMJ and motoneuron received the attention they deserved. Recently, different roles in autophagy at early and late stages of ALS were assigned due to a noncell autonomous progression of the disease in [57]. ATG7 elimination, specifically in motoneurons (ATG7 cKO) in SOD1, showed an impaired autophagy activity only in these kinds of neurons. Surprisingly, mice did not present neurodegenerative phenotypes and their motoneurons remained viable. Nevertheless, mice displayed structural and functional defects in the NMJ of some of these autophagy-truncated motoneurons, including the denervation of the tibialis anterior, which is mostly innerved by fast motoneurons. Additionally, they documented the disease progression of the SOD1G93A mouse model. They reported that only vulnerable and first-to-die motoneurons display round body aggregates that are GABARAPL1-positive at early stages, while nonvulnerable motoneurons present skeinlike aggregates that are GABARAPL1-negative at late stages. Typically, fast motoneurons die first at early stages of ALS, while slow motoneurons are more resistant and remain functional. The authors argue that fast motoneurons are able to activate autophagy machinery according to the fact that they are GABARAPL1-positive. Consequently, slow and resistant motoneurons do not recruit autophagy machinery at the same level as fast ones do. Intelligently, the authors crossed SOD1G93A and ATG7 cKO mouse models in order to explore the role of autophagy in this intriguing puzzle. As expected, fast motoneurons were affected early, resulting in the accelerated denervation of the tibialis anterior muscle. The soleus, innervated principally by slow motoneurons, remained innervated even late in the disease. The onset of tremor in this mouse model was reported earlier than in SOD1G93A due to the misfunction of these fast motoneurons. However, authors reported an extended lifespan of mice and a delay in disease progression accompanied by a reduction in glial inflammation. That is, detrimental effects of autophagy activity in motoneurons could be projected to the glia cells in the CNS in a “dying-back” communication. Several highlights can be extracted from these results: (i) selective autophagy impairment in motoneurons is not enough to trigger neurodegeneration; (ii) autophagy may be beneficial in the short term but detrimental in late stages of the disease; (iii) denervation in vulnerable motoneurons may course in a cell-autonomous way in SOD1G93A when autophagy is impaired but iv) disease progression is probably mediated by noncell autonomous mechanism—glia cells are an interesting point to be considered.
According to this, why are fast motoneurons more vulnerable than the slow ones? Fast motoneurons innerve large muscles and can be crudely divided into fatigable (FF) or resistant (FR) [58]. FF motoneurons, like those that innerve tibialis muscle, have high peak needs of ATP [59], thus, they need to maintain their mitochondria pool in good conditions to ensure the correct level of ATP. ALS patients [60] show impaired production of ATP and SOD1G93A present a low ATP/AMP ratio due to a deficient oxidative phosphorylation metabolism at early stages of the disease [61]. Recently, mitochondria located in the synapsis of spinal cord motoneurons were reported to suffer an enhancement of glucose catabolism in order to supply the inefficient production of ATP at the presymptomatic stage, while this only occurs later in the cortex motoneuron and perisynaptic glia cells [62]. An interesting hypothesis is that SOD1G93A models display deficient mitochondrial function and inadequate ATP levels. Knowing that FF motoneurons usually present a low number of mitochondria and are rapidly fatigable [63], the poor energy supply affects these motoneurons drastically due to their high metabolic demand. When autophagy is impaired, FF motoneurons are not able to degrade these defective mitochondria and provide themselves with new inputs to cellular metabolism, ending up with denervation in the short term (Figure 4). However, if autophagy is stimulated, misfunction of the NMJ is delayed, but motoneuron accumulates nonfunctional vacuolated organelles that cause earlier cell death at latest stages, as we exposed before. Accordingly, PINK1/PARKIN double KO mice are prone to accumulate defective mitochondria in the NMJ and, subsequently, cause denervation [64]. It is of note that FF motoneurons display a huge glycolytic metabolism as an energy source, thus, impaired mitochondrial ATP production may not be the unique reason for why this kind of neuron is affected first [58]. Instead, a set of features must converge in FF motoneurons to explain this selectivity. The short latency period between action potentials or the earlier endoplasmic reticulum stress are two of the possible causes [65]. Interestingly, vulnerable motoneurons display lower Ca2+ buffering compared with other kinds of neurons, which allows fast recovery times during physical exercise [66]. As a result, mitochondrion acts as an even more critical Ca2+ storage in these neurons. Since FF motoneurons show impaired Ca2+ handling at presymptomatic stages [67] and dispose of a low number of mitochondria, the accumulation of defective mitochondria may lead to a dysregulation in Ca2+ levels and consequent cell damage. Therefore, a correct balance between mitophagy and mitochondriogenesis is needed to maintain a healthy pool of these organelles and ensure an adequate intracellular concentration of Ca2+.
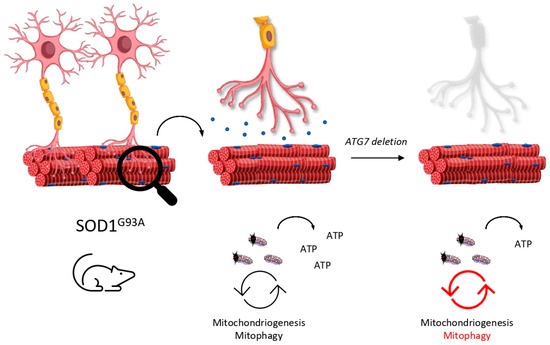
Figure 4. Mitophagy is essential to maintain the neuromuscular junction (NMJ). Inefficient renewal of mitochondria due to the selective deletion of ATG7 in motoneurons may lead to low le-vels of ATP and make them prone to denervation.
2.2.3. Mitophagy Is a Highly (Cell-Type) Dynamic Puzzle
Neuroinflammation by glial cells is recognized as an important disease modulator. Although there is still controversy about the relationship between them, autophagy stimulation is believed to reduce neuroinflammation in microglia by restricting inflammasome activation and preventing elevated ROS levels through mitophagy activity, among other processes [68]. Specifically, glial cells are broadly recognized as important modulators of ALS, which can be observed in the fact that transplanted wildtype microglia delays disease progression in ALS models [69] and the fact that selective deletion of mutant SOD1 in astrocytes affects disease progression but not its onset [70]. In the same way, selective deletion of mutant SOD1 in microglia clearly influences late stages but not early stages [71]. The work we discussed above provides the opportunity to study the course of ALS when autophagy is only limited to motoneurons, which is not possible with the use of autophagy modulators such as trehalose or rapamycin. The alleviation of neuroinflammation in the ATG7 cKO/SOD1G93A mouse model is probably due to the reduction of those vacuolated organelles that would stress neurons in the long term, but the fact that autophagy is not impaired in glial cells cannot be ignored. Therefore, could we suppose that the coelimination of autophagy activity by gene deletion in microglia and motoneurons would counteract this alleviation and induce neuroinflammation? It is a hard-to-solve question if we take into consideration that mutant SOD1 can sequester OPTN and impair mitophagy; thus, using the mutant SOD1 model implies the risk that mitophagy can be basally interfered in the control group [24]. In 2019, a heterozygous TBK1+/−/SOD1G93A mouse model was designed and studied [72]. As we exposed before, TBK1 is a key kinase that has the critical role of ensuring correct levels of mitophagy and is involved in inflammatory responses via IFN. Accordingly, TBK1+/−/SOD1G93A mice showed increased denervation in pretibial muscles and the onset of the tremor was accelerated. Moreover, at later stages, neuroinflammation was reduced, disease progression was delayed, and survival was extended. Authors argue that the deleterious autophagy/mitophagy inhibition effects can be observed at 50 days, but the beneficial impact of reducing microglia activation is overcome at 120 days. Due to the pleiotropic nature of TBK1, these data are far from revealing whether the reduction in microgliosis may correspond to either a reduction of autophagy/mitophagy activity or a decrease in neuroinflammation. Conveniently, motoneuron-selective TBK1 deletion (TBK1 cKO) was recently designed in 2020 [73]. Thus, several mouse models have been produced such as SOD1G93A, TBK1R228H/R228H/SOD1G93A (where mutated TBK1 shows no activity in any cell type), TBK1R228H/+/SOD1G93A (where TBK1 acts in a heterozygous way), and TBK1 cKO/SOD1G93A (where TBK1 is deleted only in motoneurons). According to the work discussed before, TBK1R228H/R228H/SOD1G93A and TBK1R228H/+/SOD1G93A mice present more denervation in early stages but extended survival rates compared to SOD1G93A mice. However, TBK1 cKO/SOD1G93A mice present more denervation and earlier onset of the disease but without an effect on the mice survival. This indicates that earlier denervation can be observed in a cell autonomous way when either ATG7 or TBK1 are deleted in motoneurons, but the disease progression is completed, at least in part, in a noncell autonomous way, as only noncell selective impairment of TBK1 (TBK1R228H/R228H/SOD1G93A) extends survival. Moreover, this prolonged survival in TBK1R228H/R228H/SOD1G93A is accompanied by a lower induction of a subset of interferon stimulated genes (ISGs) in glial cells, specifically in astrocytes and microglia. Therefore, neuroinflammation produced by glial cells is linked to disease progression and may be responsible for its extended lifespan (Figure 5). All the data are complemented by the fact that (i) selective-mononuclear phagocytic system deletion of ATG7 (which includes microglia) causes intestinal adhesion of macrophages, which is strongly related with inflammatory events [74], and (ii) mitophagy activity in microglia reduces neuroinflammation in Alzheimer’s disease [75]. Hence, autophagy/mitophagy in microglia may be important to ensure low levels of neuroinflammation.
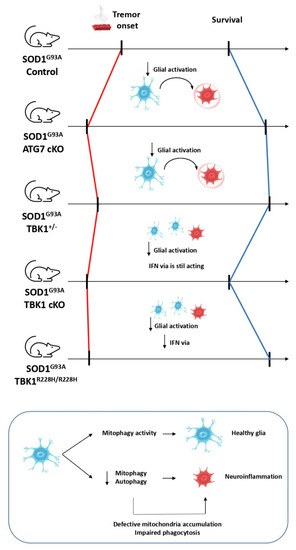
Figure 5. ALS progression presents a noncell autonomous nature. Selective deletion of autophagy genes in motoneurons causes earlier denervation in these cells. However, the SOD1 mouse model presents an extended survival, probably due to the role of glia cells in neuroinflammation.
In 2020, an alternative mitochondrial quality control was reported. It was observed that the neuroblastoma cell line SH-SY5Y, among others, releases mitochondria to extracellular space [76]. Furthermore, mitochondrial damage induced by small molecules, such as rotenone, carbonyl cyanide m-chlorophenyl hydrazone (CCCP), or mitophagy-deficient models like PARKIN mutations, increase the mitochondrial release. Extracellular mitochondria must be eliminated in order to avoid neuroinflammation, since several mitochondrial components such as cytochrome C, mitochondrial transcription factor A (TFAM), or cardiolipin can act as damage-associated molecular patterns (DAMPs) and lead to inflammatory events [77]. In parallel, a Parkinson’s disease model was documented in which defective mitochondria were released in spheroids when the dopaminergic synapsis was degenerated. Surprisingly, these mitochondria were completely degraded not in the extracellular space, but in concomitant astrocytes, where the final steps of mitophagy are performed [78]. Therefore, glial cells (at least astrocytes) show the ability to degrade these released mitochondria through a process termed as transmitophagy, in which mitochondrial degradation in neurons can be mediated by intercellular pathways. This mitochondrial delivery from the affected cell to the glia (and vice versa) has been documented not only in neurons, but also in many other cell types in the last years [79]. However, the fact that impaired mitophagy promotes this mitochondrial release opens new research fields. Phagocytosis-mediated elimination of extracellular mitochondria would end up with the delivered organelles to lysosomes, so it is not surprising that autophagy and phagocytosis share common molecular machinery as they are in charge of the elimination of intracellular and extracellular components, respectively. Indeed, a kind of endocytic pathway has been described as the LC3-associated phagocytosis (LAP), characterized by the recruitment of LC3 to the phagosome membranes involved in the phagocytosis, responsible for the degradation of several types of cargo, such as pathogens or cell debris [80]. In this context, a paradigmatic case was reported recently about microglia-selective deletion of ATG7 in a multiple sclerosis model: defective autophagy-related phagocytosis in microglia impairs myelin debris elimination. The accumulation of these remains of myelin leads to neuroinflammation and the inability to remyelinate neurons, due to the lack of ATG7 [81]. Curiously, trehalose is able to reverse the neurodegenerative phenotype in the aged mouse model because of its autophagy stimulation activity. These data raise the possibility that autophagy machinery is not only needed in microglia to avoid neuroinflammation, but is also necessary to degrade those extracellular mitochondria by glial cells (Figure 5). However, experiments in ALS models and other neurodegenerative diseases are needed to confirm this.
References
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1 Joungmok. Nat. Cell Biol. 2011, 13, 132–141.
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportuni-ties. Neuron 2017, 93, 1015–1034.
- Mizushima, N. A Brief History of Autophagy from Cell Biology to Physiology and Disease. Nat. Cell Biol. 2018, 20, 521–527.
- Lee, J.K.; Shin, J.H.; Lee, J.E.; Choi, E.J. Role of Autophagy in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2517–2524.
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; V.; Stevnsner, T.; Nilsen, H.; A.; Bohr, V.; F.; Fang, E. Mitophagy in Neurodegeneration and Aging. Neurochem. Int. 2017, 109, 202–209.
- Granatiero, V.; Manfredi, G. Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biology 2019, 8, 36.
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-Specific Autophagy in Inflammatory Diseases: A Potential Therapeutic Target Underlying the Quality Control of Multiple Organelles. Autophagy 2020, 12, 1–17.
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2016, 7, 248.
- Chen, Y.; Dorn, G.W. 2nd. PINK1- Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475.
- Weil, R.; Laplantine, E.; Curic, S.; Génin, P. Role of Optineurin in the Mitochondrial Dysfunction: Potential Implications in Neurodegenerative Diseases and Cancer. Front. Immunol. 2018, 9, 1243.
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A New Player in ALS Linking Autophagy and Neuroinflammation. Mol. Brain 2017, 10, 5.
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 534, 309–314.
- Perez, F.A.; Palmiter, R.D. Parkin-Deficient Mice Are Not a Robust Model of Parkinsonism. Proc. Natl. Acad. Sci. USA 2005, 102, 2174–2179.
- Le Grand, J.N.; Bon, K.; Fraichard, A.; Zhang, J.; Jouvenot, M.; Risold, P.Y.; Boyer-Guittaut, M.; Delage-Mourroux, R. Specific Distribution of the Autophagic Protein GABARAPL1/GEC1 in the Developing and Adult Mouse Brain and Identification of Neuronal Populations Expressing GABARAPL1/GEC1. PLoS ONE 2013, 8, e63133.
- Villa, E.; Marchetti, S.; Ricci, J.E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895.
- Chu, C.T. Mechanisms of Selective Autophagy and Mitophagy: Implications for Neurodegenerative Diseases. Neurobiol. Dis. 2019, 122, 23–34.
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C. Divergent Roles of ALS-Linked Proteins FUS/TLS and TDP-43 Intersect in Processing Long Pre-MRNAs. Nat. Neurosci. 2012, 15, 1488–1497.
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Par-kin Is a Disease Modifier in the Mutant SOD 1 Mouse Model of ALS. EMBO Mol. Med. 2018, 10, e8888.
- Chen, Y.; Deng, J.; Wang, P.; Yang, M.; Chen, X.; Zhu, L.; Liu, J.; Lu, B.; Shen, Y.; Fushimi, K.; et al. PINK1 and Parkin Are Genetic Modifiers for FUS-Induced Neurodegeneration. Hum. Mol. Genet. 2016, 25, 5059–5068.
- Cha, S.J.; Choi, H.J.; Kim, H.J.; Choi, E.J.; Song, K.H.; Im, D.S.; Kim, K. Parkin Expression Reverses Mitochondrial Dysfunc-tion in Fused in Sarcoma-Induced Amyotrophic Lateral Sclerosis. Insect Mol. Biol. 2020, 29, 56–65.
- Ghasemi, M.; Brown, R.H. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125.
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mi-tophagy That Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448.
- Sirohi, K.; Swarup, G. Defects in Autophagy Caused by Glaucoma-Associated Mutations in Optineurin. Exp. Eye Res. 2016, 144, 54–63.
- Tak, Y.J.; Park, J.H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525.
- Hadano, S.; Mitsui, S.; Pan, L.; Otomo, A.; Kubo, M.; Sato, K.; Ono, S.; Onodera, W.; Abe, K.; Chen, X.P.; et al. Functional Links between SQSTM1 and ALS2 in the Pathogenesis of ALS: Cumulative Impact on the Protection against Mutant SOD1-Mediated Motor Dysfunction in Mice. Hum. Mol. Genet. 2016, 25, 3321–3340.
- Moore, A.S.; Holzbaur, E.L.F. Dynamic Recruitment and Activation of ALS-Associated TBK1 with Its Target Optineurin Are Required for Efficient Mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358.
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a Common Pathway in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 697, 34–48.
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558.
- Huang, C.; Yan, S.; Zhang, Z. Maintaining the Balance of TDP-43, Mitochondria, and Autophagy: A Promising Therapeutic Strategy for Neurodegenerative Diseases. Transl. Neurodegener. 2020, 9, 40.
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Mo-tor-Coordinative and Cognitive Dysfunction Caused by Mutant TDP-43 Could Be Reversed by Inhibiting Its Mitochondrial Localization. Mol. Ther. 2017, 25, 127–139.
- Akizuki, M.; Yamashita, H.; Uemura, K.; Maruyama, H.; Kawakami, H.; Ito, H.; Takahashi, R. Optineurin Suppression Caus-es Neuronal Cell Death via NF-ΚB Pathway. J. Neurochem. 2013, 126, 699–704.
- Nakazawa, S.; Oikawa, D.; Ishii, R.; Ayaki, T.; Takahashi, H.; Takeda, H.; Ishitani, R.; Kamei, K.; Takeyoshi, I.; Kawakami, H.; et al. Linear Ubiquitination Is Involved in the Pathogenesis of Optineurin-Associated Amyotrophic Lateral Sclerosis. Nat. Commun. 2016, 7, 12547.
- Ahmad, L.; Zhang, S.-Y.; Casanova, J.-L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527.
- Beghi, E.; Chiò, A.; Inghilleri, M.; Mazzini, L.; Micheli, A.; Mora, G.; Poloni, M. A Randomized Controlled Trial of Recom-binant Interferon Beta-1a in ALS. Neurology 2000, 54, 469–474.
- Poutiainen, E.; Hokkanen, L.; Niemi, M.L.; Färkkilä, M. Reversible Cognitive Decline during High-Dose α-Interferon Treatment. Pharmacol. Biochem. Behav. 1994, 47, 901–905.
- Yu, J.; Zhou, X.; Chang, M.; Nakaya, M.; Chang, J.H.; Xiao, Y.; William, L.J.; Dorta-Estremera, S.; Cao, W.; Zal, A.; et al. Reg-ulation of T-Cell Activation and Migration by the Kinase TBK1 during Neuroinflammation. Nat. Commun. 2015, 6, 6074.
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491.
- Sasaki, S. Autophagy in Spinal Cord Motor Neurons in Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of Basal Autophagy in Neural Cells Causes Neurodegenerative Disease in Mice. Nature 2006, 441, 885–889.
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of Autophagy in the Central Nervous System Causes Neurodegeneration in Mice. Nature 2006, 441, 880–884.
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2430.
- Zhang, X.; Chen, S.; Lu, K.; Wang, F.; Deng, J.; Xu, Z.; Wang, X.; Zhou, Q.; Le, W.; Zhao, Y. Verapamil Ameliorates Motor Neuron Degeneration and Improves Lifespan in the SOD1G93A Mouse Model of Als by Enhancing Autophagic Flux. Aging Dis. 2019, 10, 1159–1173.
- Park, H.; Lee, J.H. Calcium Channel Blockers as Potential Therapeutics for Obesity-Associated Autophagy Defects and Fatty Liver Pathologies. Autophagy 2014, 10, 2385–2386.
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine Promotes MTOR-Independent Autophagy in the Mutant SOD1 Mouse Model of Amyotrophic Lateral Sclerosis without Slowing Dis-ease Progression. Autophagy 2018, 14, 534–551.
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Volonté, C. Actions of the Antihistaminergic Clemastine on Presymptomatic SOD1-G93A Mice Ameliorate ALS Disease Progression. J. Neuroinflammation 2016, 13, 191.
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; Van Zundert, B.; Hetz, C. Trehalose Delays the Progression of Amyotrophic Lateral Sclerosis by Enhancing Autophagy in Motoneurons. Autophagy 2013, 9, 1308–1320.
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-Independent, Autophagic Enhancer Trehalose Prolongs Motor Neuron Survival and Ameliorates the Autophagic Flux Defect in a Mouse Model of Amyotrophic Lateral Sclerosis. Autophagy 2014, 10, 588–602.
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose Decreases Mutant SOD1 Expression and Alleviates Motor Deficiency in Early but Not End-Stage Amyotrophic Lateral Sclerosis in a SOD1-G93A Mouse Model. Neuroscience 2015, 298, 12–25.
- Thau, N.; Knippenberg, S.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased MRNA Expression of PGC-1α and PGC-1αregulated Factors in the SOD1G93A ALS Mouse Model and in Human Sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074.
- Xie, Y.; Zhou, B.; Lin, M.-Y.; Wang, S.; Foust, K.D.; Sheng, Z.-H. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic FALS Mice. Neuron 2015, 87, 355–370.
- Evans, C.S.; Holzbaur, E.L.F. Lysosomal Degradation of Depolarized Mitochondria Is Rate-Limiting in OPTN-Dependent Neuronal Mitophagy. Autophagy 2020, 16, 962–964.
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium Delays Progression of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057.
- Meira Martins, L.A.; Vieira, M.Q.; Ilha, M.; de Vasconcelos, M.; Biehl, H.B.; Lima, D.B.; Schein, V.; Barbé-Tuana, F.; Borojevic, R.; Guma, F.C.R. The Interplay Between Apoptosis, Mitophagy and Mitochondrial Biogenesis Induced by Resveratrol Can Determine Activated Hepatic Stellate Cells Death or Survival. Cell Biochem. Biophys. 2014, 71, 657–672.
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786.
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal Degeneration, Distal Collateral Branching and Neuromuscular Junction Architecture Alterations Occur Prior to Symptom Onset in the SOD1G93A Mouse Model of Amyo-trophic Lateral Sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47.
- Martineau, É.; Di Polo, A.; Velde, C.V.; Robitaille, R. Dynamic Neuromuscular Remodeling Precedes Motor-Unit Loss in a Mouse Model of ALS. eLife 2018, 7, e41973.
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct Roles for Motor Neuron Autophagy Early and Late in the SOD1G93A Mouse Model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303.
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor Neuron Diversity in Development and Disease. Annu. Rev. Neurosci. 2010, 33, 409–440.
- Le Masson, G.; Przedborski, S.; Abbott, L.F. A Computational Model of Motor Neuron Degeneration. Neuron 2014, 83, 975–988.
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933.
- Ravera, S.; Bonifacino, T.; Bartolucci, M.; Milanese, M.; Gallia, E.; Provenzano, F.; Cortese, K.; Panfoli, I.; Bonanno, G. Char-acterization of the Mitochondrial Aerobic Metabolism in the Pre- and Perisynaptic Districts of the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 9220–9233.
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered Glucose Catabolism in the Presynaptic and Perisynaptic Compartments of SOD1G93A Mouse Spinal Cord and Motor Cor-tex Indicates That Mitochondria Are the Site of Bioenergetic Imbalance in ALS. J. Neurochem. 2019, 151, 336–350.
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.-S.; McNamara, J.O.; Williams, M. Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, UK, 2001.
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice. Front. Neurosci. 2017, 11, 447.
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2017, 133, 863–885.
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium Dysregulation in Amyotrophic Lateral Sclerosis. Cell Calcium 2010, 47, 165–174.
- Barrett, E.F.; Barrett, J.N.; David, G. Dysfunctional Mitochondrial Ca2+ Handling in Mutant SOD1 Mouse Models of FALS: Integration of Findings from Motor Neuron Somata and Motor Terminals. Front. Cell. Neurosci. 2014, 8, 184.
- Strohm, L.; Behrends, C. Glia-Specific Autophagy Dysfunction in ALS. Semin. Cell Dev. Biol. 2020, 99, 172–182.
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-Type Microglia Extend Survival in PU.1 Knockout Mice with Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026.
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleve-land, D.W. Astrocytes as Determinants of Disease Progression in Inherited Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2008, 11, 251–253.
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392.
- Brenner, D.; Sieverding, K.; Bruno, C.; Lüningschrör, P.; Buck, E.; Mungwa, S.; Fischer, L.; Brockmann, S.J.; Ulmer, J.; Bliederhäuser, C.; et al. Heterozygous Tbk1 Loss Has Opposing Effects in Early and Late Stages of ALS in Mice. J. Exp. Med. 2019, 216, 267–278.
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 2020, 106, 789–805.e5.
- Lee, Y.; Lee, J.W.; Nam, H.; Yu, S.W.; Yu, S.W. Cx3cr1 CreERT2-Driven Atg7 Deletion in Adult Mice Induces Intestinal Adhe-sion. Mol. Brain 2020, 13, 88.
- Lautrup, S.; Lou, G.; Aman, Y.; Nilsen, H.; Tao, J.; Fang, E.F. Microglial Mitophagy Mitigates Neuroinflammation in Alz-heimer’s Disease. Neurochem. Int. 2019, 129, 104469.
- Choong, C.-J.; Okuno, T.; Ikenaka, K.; Baba, K.; Hayakawa, H.; Koike, M.; Yokota, M.; Doi, J.; Kakuda, K.; Takeuchi, T.; et al. Alternative Mitochondrial Quality Control Mediated by Extracellular Release. Autophagy 2020, 1–13, doi:10.1080/15548627.2020.1848130.
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroin-flammation. Mediat. Inflamm. 2019, 2019, 4050796.
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial Transmi-tophagy and Parkinson’s Disease. Glia 2020, 68, 2277–2299.
- Jackson, J.G.; Robinson, M.B. Regulation of Mitochondrial Dynamics in Astrocytes: Mechanisms, Consequences, and Un-knowns. Glia 2018, 66, 1213–1234.
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598.
- Berglund, R.; Guerreiro-Cacais, A.O.; Adzemovic, M.Z.; Zeitelhofer, M.; Lund, H.; Ewing, E.; Ruhrmann, S.; Nutma, E.; Parsa, R.; Thessen-Hedreul, M.; et al. Microglial Autophagy-Associated Phagocytosis Is Essential for Recovery from Neuroin-flammation. Sci. Immunol. 2020, 5, eabb5077.


