 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | José I. Jiménez-Zurdo | + 787 word(s) | 787 | 2020-03-17 02:53:35 | | | |
| 2 | Nicole Yin | Meta information modification | 787 | 2020-03-25 04:02:06 | | | | |
| 3 | Nicole Yin | Meta information modification | 787 | 2020-03-25 10:14:24 | | | | |
| 4 | Nicole Yin | Meta information modification | 787 | 2020-03-25 10:24:39 | | | | |
| 5 | Nicole Yin | + 3 word(s) | 790 | 2020-10-27 07:56:12 | | |
Video Upload Options
It is now widely recognized that genetic regulation of bacterial physiology cannot be fully understood without considering the possible participation of small non-coding RNAs (sRNAs). However, current genome annotations for most bacterial species unaccountably overlook sRNA genes. Here, we describe strategies undertaken to characterize the noncoding transcriptome of rhizobia, a group of soil bacteria that are well-known for their ability to establish agriculturally and environmentally relevant mutualistic nitrogen-fixing symbioses with legumes.
1. Introduction
Small non-coding RNAs (sRNAs) are ubiquitous components of bacterial adaptive regulatory networks underlying stress responses and intracellular infection of eukaryotic hosts.
2. Deciphering the Noncoding Transcriptome of Nitrogen-Fixing Legume Symbionts
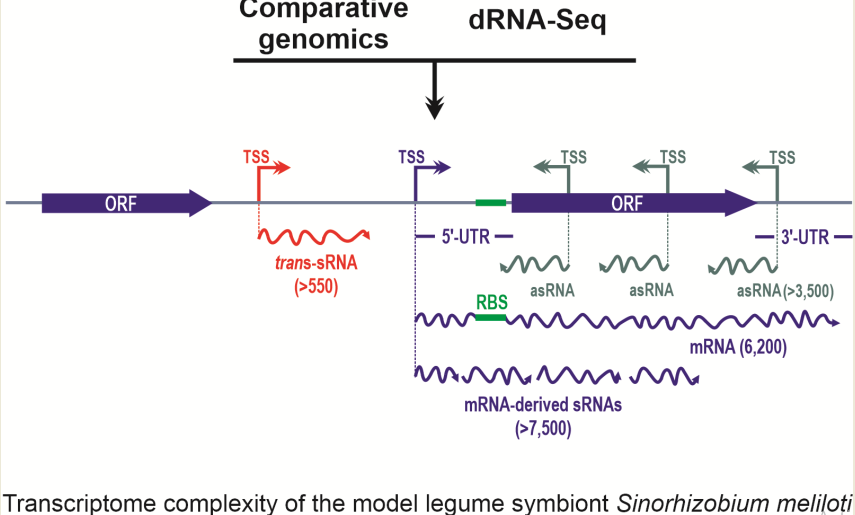
However, non-protein coding genes largely escape classical genetics screens and the primary annotation of a single bacterial genome sequence, which is essentially limited to the prediction of open reading frames (ORFs), tRNAs, and rRNAs. Before the advent of high-throughput sequencing, computational comparative genomics was thus the tool of choice to identify conserved regions with putative functions in the unannotated portions of the genome. Accordingly, pioneering genome-wide searches for sRNAs in rhizobia relied on the comparison of intergenic sequences (i.e., genomic regions between ORFs; IGRs) from phylogenetically close species to unveil trans-acting sRNAs. Specifically, three studies published almost concurrently used the IGRs from the alfalfa symbiont S. meliloti (reference strain Rm1021) as queries to interrogate the genomes of α–proteobacterial relatives, i.e., plant symbionts (e.g., Mesorhizobium loti, Rhizobium etli or R. leguminosarum bv. viciae), phytopathogens (Agrobacterium tumefaciens) and animal pathogens (Brucella species) [1][2][3]. All three searches combined genomic comparisons with other known features of trans-sRNAs, namely association with orphan transcription signatures (promoter motifs and/or Rho-independent transcriptional terminators) and conservation of thermodynamically stable secondary structures. Collectively, these approaches predicted more than a hundred IGRs in the three replicons (chromosome, and pSymA and pSymB megaplasmids) of the Rm1021 genome putatively encoding a trans-sRNA. Northern blot hybridization of total RNA, RACE (Rapid Amplification of cDNA Ends) mapping of transcripts boundaries and/or microarray probing experimentally confirmed that a few dozens of these candidate IGRs did express sRNA species from independent transcription units. These studies also anticipated the symbiotic and/or stress-dependent expression of subsets of the identified sRNAs. A similar combination of in silico searches and experimental approaches also delivered the first inventories of sRNAs expressed by R. etli CFN42, Bradyrhizobium japonicum USDA110 and M. huakuii 7653R [4][5][6], the symbiotic partners of common bean (Phaseolus vulgaris), soybean (Glycine max), and milkvetch (Astragalus sinicus), respectively.
Inherent limitations to both computational searches and microarray designs necessarily biased these seminal genome-wide screens for sRNAs in rhizobia to the identification of putative intergenic, trans-acting, conserved riboregulators. Straightforward experimental identification of transcription start sites (TSS) associated to coding sequences, untranslated mRNA regions, and noncoding RNA genes in the prokaryotic genomes is now feasible with the implementation of oriented differential RNASeq (dRNASeq) [7] or Cappable-Seq [8] strategies. These two experimental setups target the primary transcriptome on a strand-specific basis upon terminal exonuclease (TEX)-mediated depletion of the processed RNA species or streptavidin capture of primary transcripts capped at their distinctive triphosphorylated 5’-ends, respectively. In particular, dRNASeq surveys rediscovered the early-identified sRNAs in S. meliloti and B. japonicum and further uncovered the complex rhizobial transcriptomes with the addition of hundreds of unknown trans-sRNAs, as well as thousands of mRNA-derived sRNAs and antisense RNAs (asRNAs) [5][9][10][11][12]. Other RNASeq studies are conceived to profile specific subpopulations of cellular transcripts supposedly enriched in sRNAs, e.g., RNA species co-immunoprecipitated with the major bacterial RNA chaperone Hfq. However, this approach resulted in a minor addition to the sRNA landscape revealed by deep sequencing of S. meliloti total RNA[13].
Prokaryotic gene prediction pipelines such as EuGen-P have incorporated the novel gene structural features uncovered by dRNASeq for the accurate reannotation of the S. meliloti (strains Rm1021 and Rm2011) and B. japonicum USDA110 genomes[10][11][12]. Even though dRNASeq mostly serves annotation purposes, comparison of transcripts levels in some datasets identified differentially expressed sRNAs in free-living and nodule endosymbiotic bacteria. In this regard, it is noteworthy the identification of nodule-expressed sRNAs by RNASeq based profiling of RNA derived from each developing zone of indeterminate nodules induced on the model legume M. truncatula by Rm2011[14]. On the other hand, comprehensive mapping of TSS in Rm2011 and USDA110 has facilitated the prediction of motifs putatively recognized by alternative σ factors such as RpoE2 (σE2) or RpoN (σ54) in the promoter regions of some of the identified sRNAs, thus placing these RNA regulators in major stress response and/or symbiotic regulons[10][11][12]. The integration of the updated genome annotation files, primary expression profiles, and promoter predictions provides a solid resource for the forthcoming investigation of the function of sRNAs in plant symbiotic bacteria. Similar in silico and experimental workflows can be implemented to explore the noncoding transcriptome of any bacterial species.
References
- Vincent M Ulvé; Emeric W Sevin; Angélique Chéron; Frédérique Barloy-Hubler; Identification of chromosomal alpha-proteobacterial small RNAs by comparative genome analysis and detection in Sinorhizobium meliloti strain 1021. BMC Genomics 2007, 8, 467-467, 10.1186/1471-2164-8-467.
- Claudio Valverde; Jonathan Livny; Jan-Philip Schlüter; Jan Reinkensmeier; A Becker; Gustavo Parisi; Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011. BMC Genomics 2008, 9, 416-416, 10.1186/1471-2164-9-416.
- Coral Del Val; Elena Rivas; Omar Torres-Quesada; Nicolás Toro; José I. Jiménez-Zurdo; Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Molecular Microbiology 2007, 66, 1080-1091, 10.1111/j.1365-2958.2007.05978.x.
- Maarten Vercruysse; Maarten Fauvart; Lore Cloots; Kristof Engelen; Inge M Thijs; Kathleen Marchal; Jan Michiels; Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genomics 2010, 11, 53-53, 10.1186/1471-2164-11-53.
- Ramakanth Madhugiri; Gabriella Pessi; Björn Voß; Julia Hahn; Cynthia M. Sharma; Richard Reinhardt; Jörg Vogel; Wolfgang R Hess; Hans-Martin Fischer; Elena Evguenieva-Hackenberg; et al. Small RNAs of the Bradyrhizobium/Rhodopseudomonas lineage and their analysis. RNA Biology 2012, 9, 47-58, 10.4161/rna.9.1.18008.
- Xie Fuli; Zhao Wenlong; Wang Xiao; Zhang Jing; Hao Baohai; Zou Zhengzheng; Ma Bin-Guang; Youguo Li; A Genome-Wide Prediction and Identification of Intergenic Small RNAs by Comparative Analysis in Mesorhizobium huakuii 7653R. Frontiers in Cellular and Infection Microbiology 2017, 8, 1730, 10.3389/fmicb.2017.01730.
- Cynthia M. Sharma; Jörg Vogel; Differential RNA-seq: the approach behind and the biological insight gained. Current Opinion in Microbiology 2014, 19, 97-105, 10.1016/j.mib.2014.06.010.
- Laurence M. Ettwiller; John Buswell; Erbay Yigit; Ira Schildkraut; A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome.. BMC Genomics 2016, 17, 199, 10.1186/s12864-016-2539-z.
- Jan-Philip Schlüter; Jan Reinkensmeier; Svenja Daschkey; Elena Evguenieva-Hackenberg; Stefan Janssen; Sebastian Jaenicke; Jörg Becker; Robert Giegerich; Anke Becker; A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 2010, 11, 245-245, 10.1186/1471-2164-11-245.
- Jan-Philip Schlüter; Jan Reinkensmeier; Melanie J Barnett; Claus Lang; Elizaveta Krol; Robert Giegerich; Sharon R. Long; Anke Becker; Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 2013, 14, 156-156, 10.1186/1471-2164-14-156.
- Jelena Čuklina; Julia Hahn; Maxim V. Imakaev; Ulrich Omasits; Konrad U. Förstner; Nikolay Ljubimov; Melanie Goebel; Gabriella Pessi; Hans-Martin Fischer; Christian H. Ahrens; et al.M.S. GelfandElena Evguenieva-Hackenberg Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis - a rich resource to identify new transcripts, proteins and to study gene regulation.. BMC Genomics 2016, 17, 302, 10.1186/s12864-016-2602-9.
- Erika Sallet; Brice Roux; Laurent Sauviac; Marie-Francoise Jardinaud; Sébastien Carrère; Thomas Faraut; Fernanda De Carvalho-Niebel; Jérôme Gouzy; Pascal Gamas; Delphine Capela; et al.Claude BruandThomas Schiex Next-generation annotation of prokaryotic genomes with EuGene-P: application to Sinorhizobium meliloti 2011.. Current Neuropharmacology 2013, 20, 339-54, 10.1093/dnares/dst014.
- Omar Torres-Quesada; Jan Reinkensmeier; Jan-Philip Schlüter; Marta Robledo; Alexandra Peregrina; Robert Giegerich; Nicolás Toro; Anke Becker; José I. Jiménez-Zurdo; Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biology 2014, 11, 563-579, 10.4161/rna.28239.
- Brice Roux; Nathalie Rodde; Marie-Francoise Jardinaud; Ton Timmers; Laurent Sauviac; Ludovic Cottret; Sébastien Carrère; Erika Sallet; Emmanuel Courcelle; Sandra Moreau; et al.Frédéric DebelléDelphine CapelaFernanda De Carvalho-NiebelJérôme GouzyClaude BruandPascal GamasFernanda Carvalho‐Niebel An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. The Plant Journal 2014, 77, 817-837, 10.1111/tpj.12442.


