 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zichao Li | + 2251 word(s) | 2251 | 2021-02-03 08:50:49 | | | |
| 2 | Vicky Zhou | Meta information modification | 2251 | 2021-02-05 02:47:52 | | |
Video Upload Options
Autophagy, which is a conserved biological process and essential mechanism in maintaining homeostasis and metabolic balance, enables cells to degrade cytoplasmic constituents through lysosomes, recycle nutrients, and survive during starvation. Autophagy exerts an anticarcinogenic role in normal cells and inhibits the malignant transformation of cells. On the other hand, aberrations in autophagy are involved in gene derangements, cell metabolism, the process of tumor immune surveillance, invasion and metastasis, and tumor drug-resistance. Therefore, autophagy-targeted drugs may function as anti-tumor agents. Accumulating evidence suggests that flavonoids have anticarcinogenic properties, including those relating to cellular proliferation inhibition, the induction of apoptosis, autophagy, necrosis, cell cycle arrest, senescence, the impairment of cell migration, invasion, tumor angiogenesis, and the reduction of multidrug resistance in tumor cells. Flavonoids, which are a group of natural polyphenolic compounds characterized by multiple targets that participate in multiple pathways, have been widely studied in different models for autophagy modulation. However, flavonoid-induced autophagy commonly interacts with other mechanisms, comprehensively influencing the anticancer effect. Accordingly, targeted autophagy may become the core mechanism of flavonoids in the treatment of tumors.
1. Introduction
1.1. Flavonoids
Flavonoids, as naturally occurring polyphenols, are widely present in a variety of plants employed for both medicine and food [1]. In ancient times, they were considered of great medicinal value and established a vital therapeutic effect in the medicinal system [2]. Flavonoids exhibit potent activities in the chemoprevention and treatment of many chronic human diseases, such as tumors, possessing an inestimable development potential.
Emerging evidence suggests that the anticarcinogenic mechanisms of flavonoids include proliferation inhibition, the induction of apoptosis, autophagy, necrosis, cell cycle arrest, senescence, the impairment of cell migration, invasion, tumor angiogenesis, and the reduction of multidrug resistance in tumor cells [2][3][4][5]. In addition, flavonoids have a cytoprotective or cytotoxic effect on tumor cells through acting with multiple targets participating in multiple pathways associated with the development of cancer. For example, protective autophagy induced by flavonoids resists its anti-tumor effects, while autophagic cell death induced by flavonoids exerts a synergistic anti-tumor effect. Flavonoids serve as chemo-preventive and therapeutic agents for diverse cancers through manipulating autophagy. For instance, Apigenin has a preventive effect on UV-induced skin cancer by apigenin-induced autophagy through stimulating AMP-activated protein kinase (AMPK) or inhibiting protein kinase B (AKT) activation in keratinocytes (both human and mouse keratinocyte cell lines and primary normal human epidermal keratinocytes) to inhibit UV-mediated mammalian target of rapamycin (mTOR) activation [6][7].
Autophagy, which is the pathway of cell death induced by flavonoids, is acknowledged to be an effective strategy for the development and improvement of anticancer drugs [3][8]. Autophagic signals commonly form crosstalk with other mechanisms, comprehensively creating a complex network.
1.2. Autophagy Mechanisms
Autophagy, which is a common life phenomenon and an important cell survival mechanism in eukaryotes, can maintain cell metabolism and homeostasis under stress by capturing and degrading its own redundant and damaged organelles, as well as cytoplasmic protein aggregates. It is mainly divided into three types: Macroautophagy; microautophagy; and chaperone-mediated autophagy. Macroautophagy is a metabolic process in which cells wrap proteins or organelles to form autophagososomes through bilayer membranes. Then, the outer membrane fuses with the lysosomal membrane to form autophagolysosomes, and finally degrades the wrapped contents by hydrolyzing enzymes. The other two types of autophagy are just different in their mode of delivering the particular cargo to be degraded by lysosomes [9], which will not be discussed in this article.
In mammals, more than 30 autophagy-related genes (ATGs) have been discovered, and they are mainly responsible for the formation of functional complexes which serve as the core pathway of autophagy [10]. When cells enter into a state of stress, silencing key ATG genes, such as ATG3, ATG4, Beclin1/ATG6, ATG10, and ATG12, can induce tumor transformation [11]. Homologs of Atg1 (unc-51-like kinase (ULK)1 or ULK2), ATG13, and scaffold protein FAK interacting protein 200 kD (FIP200, the homologous protein of yeast ATG17) form ULK complexes, receiving signals from nutritional conditions and recruiting downstream Atg proteins participating in autophagosome formation [11][12]. In the case of nutritional deficiency, the inactivation of rapamycin complex mTORC1 can induce activation of the unc-51-like kinase (ULK1/2) complex, which phosphorylates FIP200 in the process of autophagy initiation [13]. In addition, mTOR, as the main negative regulator of autophagy in cancer cells, responds to specific signals between cell growth and autophagy, such as the nutritional status, growth factors, stress, and so on [11]. Subsequently, the activated ULK complex phosphorylates the PI3K-Beclin1 protein complex (composed of Beclin1, ATG14L, p150, and Vps34), which induces nucleation and initial autophagosomal membrane formation [12][14]. Additionally, the complex recruits two interrelated ubiquitin-like protein (UBL) binding systems to regulate membrane elongation and autophagosome expansion [15]. Autophagy-associated proteins ATG5 and ATG12 interact with ATG16 to form AG12-ATG5-AG16 complexes with an E3-like enzyme function. Simultaneously, ATG4 hydrolyzes LC3 to produce the LC3-I precursor molecule, which binds to E1 ligase (ATG7) and transfers to E2 ligase (ATG3). Finally, the ATG12-ATG5-ATG16L1 complex promotes the coupling of LC3-I with phosphatidylethanolamine (PE), resulting in LC3-II remaining on mature autophagosomes to end the elongation reaction [14][16]. In addition, the LC3-II left on the autophagosome can recruit a selective substrate by binding to the autophagy receptor protein p62/SQSTM (P62). Once the two bind to the LC3-II protein, part of the protein is recycled into LC3-I under the action of Atg4B [17]. Acid lysosomes combine with autophagosomes to form autophagolysosomes and then degrade the contents. The specific mechanism is shown in Figure 1.
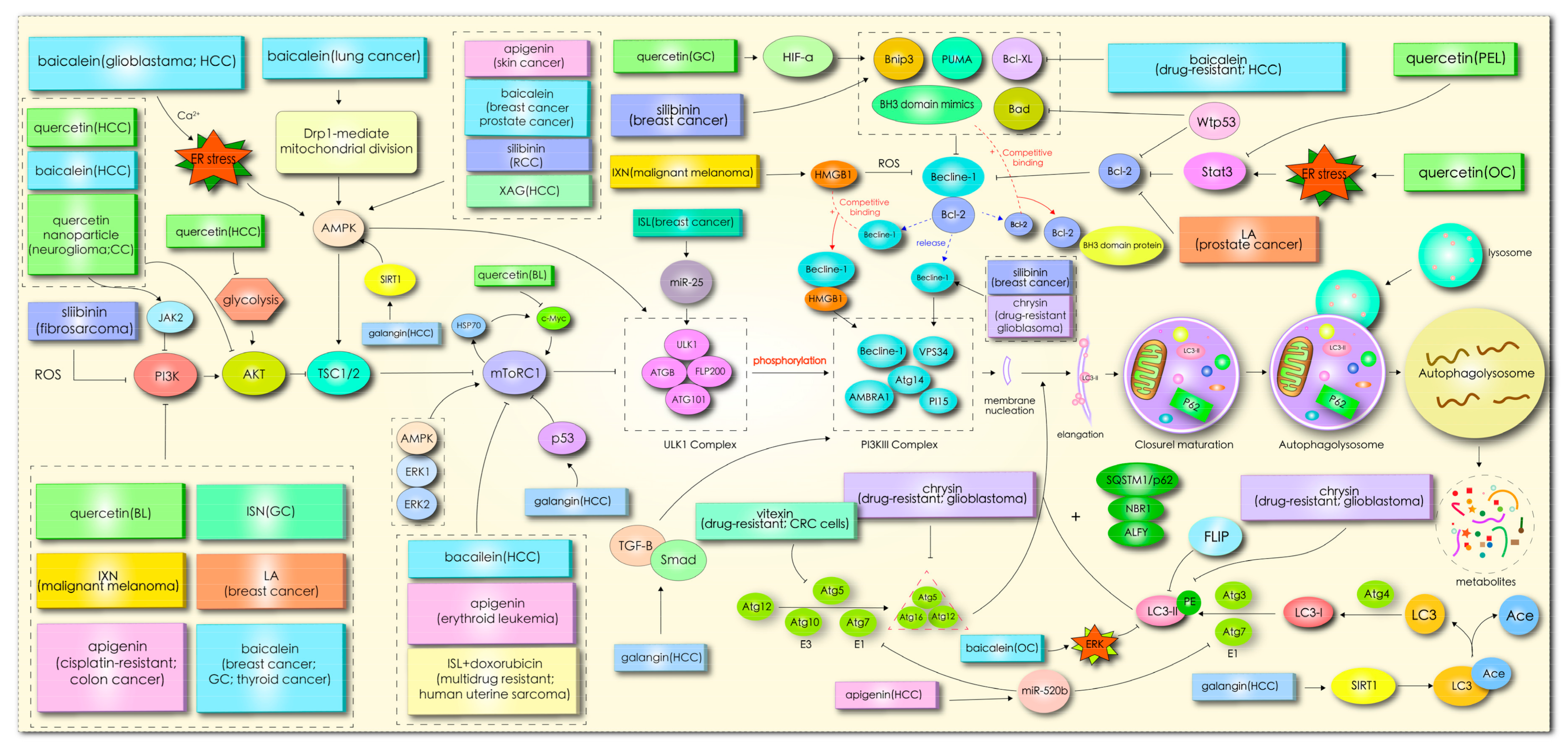
1.3. Role of Autophagy in Cancer
There is a complicated relationship between the changes associated with autophagy and therapy from the development of a tumor, depending on the cancer type [18]. Autophagy plays different roles in different cells, external factors of the same cell, or stages of tumor development [19]. The baseline level of autophagy in normal cells is relatively low, yet there are inconsistent levels of autophagy in various tumor cells [20]. Studies have shown that the autophagy level in certain tumor cells is lower than in normal cells, even in the case of hypoxia or nutritional deficiency, and the autophagy level cannot be enhanced due to monoallelic deletion of the BECN1 gene, such as in breast cancer, ovarian cancer, and prostate cancer cells; however, for most tumor cells or advanced cancer cells, a higher level of basic autophagy is needed to meet the higher metabolic demands [20][21][22].
In normal cells, the functions of autophagy include the removal of abnormally folded proteins and damaged organelles for balancing energy sources, the prevention of DNA damage, a reduction of the cellular stress response, and tumor incidence. However, in cancers, this function of autophagy plays the opposite role, which may facilitate the survival of tumor cells [20]. Tumor cells suffer from various metabolic pressures in the process of growth, from hypoxia to nutritional deficiency, and even chemotherapy intervention, while the existence of autophagy means that normal cells or tumor cells have metabolic plasticity, so that cells can adapt to a series of harsh environments [9][23]. For example, in the model of fibroblast-breast cancer cell MCF-7 co-culture for simulating the tumor microenvironment, autophagy induction could reduce the mitochondrial mass in fibroblasts, resulting in aerobic respiratory inactivation and glycolysis conversion to provide rich metabolites (lactic acid and pyruvate) supporting the oxidative phosphorylation of tumor cells [24]. Furthermore, autophagy functions as a tumor promoter through participation in diverse metabolic pathways to degrade various substrates and provide fuel for almost all aspects of carbon metabolism, such as the degradation of carbohydrates into sugars and DNA into nucleosides to promote glycolysis, and the conversion of proteins into amino acids and lipids into fatty acids to facilitate the tricarboxylic acid (TCA) cycle [9]. Apparently, autophagy suppression during tumor progression can destroy tumor metabolism, leading to adverse metabolic consequences, including impaired mitochondrial metabolism, cholesterol accumulation, redox imbalance, nucleotide pool depletion, and a reduced energy charge; in some cases, it may even lead to impaired tumor growth and tumor cell death [9].
Autophagy has an undesirably complicated role in promoting tumor initiation, growth, survival, deterioration, and metastasis. At the early stage of the tumor, autophagy restrains tumorigenesis and silencing autophagy-related genes will lead to DNA damage and genomic instability, triggering initial tumorigenesis [19]. In certain tumors, such as those caused by chronic tissue injury or inflammation, autophagy knockout increases tumor initiation in the pancreas and liver of mice. However, autophagy is still required for the tumor to develop to the malignant stage. Therefore, autophagy removal is not conducive to the growth of malignant tumors, but contributes to the formation of benign tumors [23].
2. Autophagy, Flavonoids and Cancer
Abnormal autophagy has been shown to be related to the malignant phenotype and a poor prognosis of human cancer, but the detailed mechanism is still unclear. The role of autophagy in cancer is context-dependent, inhibiting the growth of cancer cells in the early stage of tumorigenesis and promoting tumor survival in advanced malignant tumors [25]. Understanding the dependence of autophagy on the cell type specificity will help to clarify the mechanism of autophagy in tumor cells. In most tumor cells, highly expressed autophagy-associated proteins and signaling pathways are activated to coordinate autophagy and promote tumor cell growth and survival. The drug inhibition of autophagy is expected to be an effective anticancer strategy. Some small autophagy inhibitors have been studied and used in the treatment of several diseases, such as PI3K inhibitors (3-MA, wortmannin, and LY294002), mTOR inhibitors (rapamycin and its analogues, RAD001, CCI-779, and Deforolimus), an autophagososome formation inhibitor (desmethylclomipramine), autophagolysosome formation inhibitors (CQ, bafilomycin A1, and thapsigargin), etc., but these inhibitors are well-tolerated and have limited or no curative effect [26]. All in all, the lack of a molecular target in these inhibitors and concerns about the adaptability of the system in the body underscore the need to develop more potent, specific, and translatable inhibitors of autophagy. Low-toxicity natural compounds (such as flavonoids), especially those of a dietary origin, are receiving increasing attention in the prevention and treatment of diseases, including cancer. Elucidating that flavonoid-induced autophagy promotes or inhibits cell survival may provide a new perspective for clinical research on flavonoids that may circumvent drug resistance by targeting protective autophagy [27].
The above summary shows that different flavonoids play different roles in various types of cancer cells, which can be regarded as autophagy inhibitors and autophagy inducers. Silibinin-induced autophagy has toxic effects on cells, such as inducing autophagic cell death, inhibiting cell invasion and migration, and overcoming drug resistance. Quercetin and EGCG mainly induce protective autophagy and play a protective role in promoting tumor growth, even in many human diseases. In addition, quercetin and luteolin also inhibit the occurrence of hunger-induced protective autophagy in glioblastoma. Chalcone compounds also exert an anti-tumor effect by targeting tumor cell autophagy; although there are not many studies at present, it has partly been shown that autophagy can be used as a therapeutic target of chalcone compounds in tumor cytochemotherapy. For example, isoliquiritigenin inhibits autophagy to enhance the sensitivity of drug-resistant uterine sarcoma cells to doxorubicin, and induces autophagy and epirubicin to promote the death of drug-resistant breast cancer cells; hydroxysafflor yellow A-mediated autophagy suppresses the proliferation of hepatoma cells; and XAG-triggered autophagy contributes to the anti-metastatic capacity of hepatoma cells. Interestingly, flavonoids overcome MDR by regulating autophagy induced by chemotherapeutic drugs, irrespective of whether they induce protective autophagy or autophagic cell death in non-drug-resistant cells. The possible reason for this is that the pathway of autophagy mediated by flavonoids is different from that of chemotherapeutic drugs. Taken together, the combined use of antineoplastic chemotherapeutic drugs and flavonoids will help to solve the common problem of multidrug resistance of chemotherapeutic drugs. Although it has been preliminarily confirmed in vivo and in vitro that the combination of flavonoids and chemotherapeutic drugs plays a role through targeted autophagy, clinical trial data are currently insufficient.
After different types of cells are treated with flavonoids, autophagy activated by the kinase signal pathway or transcription factors may be simultaneously involved in the regulation of cell growth and proliferation, apoptosis, necrosis, cell cycle arrest, senescence, cell migration, invasion, tumor angiogenesis, and multidrug resistance.
The effect of flavonoids on autophagy were partly regulated by signal pathway PI3K/AKT/mTOR, as shown in Figure 1. This demonstrates the importance and universality of this signal pathway in affecting the autophagy of flavonoids. An abnormal expression of the PI3K/Akt gene regulates the exchange of autophagy activity. In the initial stage of autophagy, PI3K, AKT, AMPK, mTOR, ULK1, and beclin1 kinases are implicated in the regulation of autophagy, while flavonoids coordinate the expression of these kinases through ER stress, ROS, p53, Bcl-2, microRNA, HMGB1, glycolysis, and other pathways to affect autophagy. In this review, it was found that the current studies on flavonoid-mediated autophagy are mainly focused on the initial stage of autophagy membrane formation, while there are few studies on the late extension, maturation, and fusion stage, which may represent another direction of flavonoid targeted autophagy research. Flavonoids affect the occurrence and development of tumor cells by stimulating autophagy signals, so targeted autophagy may become the core approach of flavonoids in the treatment of tumors.
In summary, activation of the autophagy signal plays a vital role in the anti-tumor effect of flavonoids, especially the interaction with other signal pathways. However, the clinical application of flavonoids is limited due to their diverse structure, numerous binding sites, and complex action mechanisms. In addition, it is also necessary to strengthen in-depth research, such as structure–activity relationship research, so that flavonoids can be used as leading compounds for structural modification and structural optimization to develop a new generation of drugs.
References
- Chahar, M.; Sharma, N.; Dobhal, M.; Joshi, Y.J.P.R. Flavonoids: A versatile source of anticancer drugs. Pharmacogn. Rev. 2011, 5, 1–12.
- Wang, S.; Wu, M.; Cai, C.; Li, M.; Lu, J.J.J.O.E. Autophagy modulators from traditional Chinese medicine: Mechanisms and therapeutic potentials for cancer and neurodegenerative diseases. J. Ethnopharmacol. 2016, 194, 861–876.
- Fang, D.; Xiong, Z.; Xu, J.; Yin, J.; Luo, R. Chemopreventive mechanisms of galangin against hepatocellular carcinoma: A review. Biomed. Pharmacother 2019, 109, 2054–2061.
- Liao, C.; Lee, C.; Tsai, C.; Hsueh, C.; Wang, C.; Chen, I.; Tsai, M.; Liu, M.; Hsieh, A.; Su, K.; et al. Novel Investigations of Flavonoids as Chemopreventive Agents for Hepatocellular Carcinoma. BioMed Res. Int. 2015, 2015, 840542.
- Tong, X.; Bridgeman, B.B.; Smith, K.A.; Avram, M.J.; Pelling, J.C. AMPK-mTOR axis as key target for chemoprevention of UV-induced skin cancer by the bioflavonoid apigenin. Cancer Res. 2012, 72.
- Bridgeman, B.B.; Wang, P.; Ye, B.; Pelling, J.C.; Volpert, O.V.; Tong, X. Inhibition of mTOR by apigenin in UVB-irradiated keratinocytes: A new implication of skin cancer prevention. Cell Signal. 2016, 28, 460–468.
- Kiruthiga, C.; Devi, K.; Nabavi, S.; Bishayee, A.J.C. Autophagy: A Potential Therapeutic Target of Polyphenols in Hepatocellular Carcinoma. Cancers 2020, 12, 562.
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043.
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435.
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295.
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139.
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003.
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372.
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896.
- Kumanomidou, T.; Mizushima, T.; Komatsu, M.; Suzuki, A.; Tanida, I.; Sou, Y.-S.; Ueno, T.; Kominami, E.; Tanaka, K.; Yamane, T. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 2006, 355, 612–618.
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158.
- Yang, X.; Yu, D.-D.; Yan, F.; Jing, Y.-Y.; Han, Z.-P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.-X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14.
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967.
- Hippert, M.M.; O’Toole, P.S.; Thorburn, A. Autophagy in cancer: Good, bad, or both? Cancer Res. 2006, 66, 9349–9351.
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930.
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619.
- Martinez-Outschoorn, U.; Balliet, R.; Rivadeneira, D.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.; Lin, Z.; Witkiewicz, A.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276.
- Zhang, S.-W.; Feng, J.-N.; Cao, Y.; Meng, L.-P.; Wang, S.-L. Autophagy prevents autophagic cell death in Tetrahymena in response to oxidative stress. Zool. Res. 2015, 36, 167–173.
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153.
- Zhang, L.; Cheng, X.; Gao, Y.; Zheng, J.; Xu, Q.; Sun, Y.; Guan, H.; Yu, H.; Sun, Z. Apigenin induces autophagic cell death in human papillary thyroid carcinoma BCPAP cells. Food Funct. 2015, 6, 3464–3472.
- Sengupta, A.; Molkentin, J.D.; Yutzey, K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 2009, 284, 28319–28331.
- Wang, K.; Liu, R.; Li, J.; Mao, J.; Lei, Y.; Wu, J.; Zeng, J.; Zhang, T.; Wu, H.; Chen, L.; et al. Quercetin induces protective autophagy in gastric cancer cells: Involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy 2011, 7, 966–978.


