 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alessandro Parodi | + 3787 word(s) | 3787 | 2021-01-24 13:52:15 | | | |
| 2 | Peter Tang | Meta information modification | 3787 | 2021-02-06 10:32:21 | | |
Video Upload Options
Cysteine cathepsins are lysosomal enzymes belonging to the papain family. Their expression is misregulated in a wide variety of tumors, and ample data prove their involvement in cancer progression, angiogenesis, metastasis, and in the occurrence of drug resistance.
1. Introduction
Cathepsins are lysosomal proteases and, according to their active site, they can be classified into cysteine, aspartate, and serine cathepsins [1]. Human cysteine cathepsins (Cts) are ubiquitously present in all organisms ranging from prokaryotes to mammals and have a highly conserved residue of cysteine in their active site. They are essential in the degradation of the proteins that are internalized in the lysosomes through endocytosis, phagocytosis, and autophagocytosis [2]. Furthermore, they are involved in the cellular protein digestion, zymogen activation, and extracellular matrix (ECM) remodeling [3][4]. Under physiological conditions, Cts are fundamental in maintaining tissue homeostasis, and they are involved in different processes such as immune response, apoptosis, development, and differentiation [5]. Alterations in Cts expression, localization, and activity have been associated with several pathological disorders, including cancer progression [6][7], and their ectopic expression is usually associated with poor prognosis [8].
Recent data have demonstrated that Cts localization is not limited to the endolysosomal compartment, but they were also found in the cell cytoplasm, nucleus, mitochondria, and extracellular space, indicating their broad biological activity [9][10][11]. In the context of cancerogenesis, secreted Cts contribute to the tumor ECM degradation and remodeling, while intracellular cathepsins are pivotal components of signaling pathways, which can enhance cancer cell growth and inflammation [12][13]. Also, Cts are engaged in response to anticancer therapy within the tumor microenvironment, and they can have crucial roles in the development of resistance phenomena to the therapeutics [14][15][16].
2. Cts Synthesis, Structure, and Localization
The family of Cts proteases includes the subtypes B, C, F, H, K, L, O, S, V, W, and X [17], which are synthesized as inactive zymogens and processed into their mature forms in the acidic environment of the lysosomes. In addition to the pH and the redox status of the surrounding microenvironment, their proteolytic activity is regulated by biological activators and inhibitors, such as cytokines, growth factors, collagen peptides, and endogenous inhibitors [18][19].
According to their proteolytic activity, they can be further classified into exopeptidases (Cts C and X) or endopeptidases (F, O, S, K, V, L, and W), with Cts H and B possessing both endo- and exopeptidase activities (Figure 1a) [20].
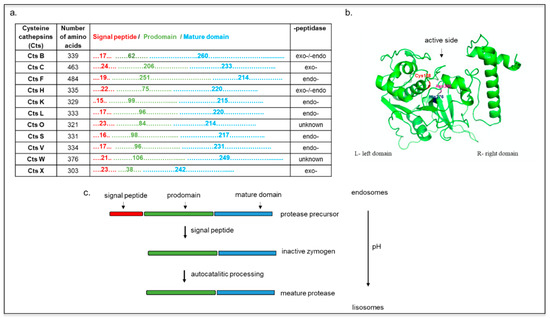
Figure 1. (a) Cysteine cathepsins (Cts) functional sequences. Representation of Cts according to their number of amino acids, length of domains (signal peptide, prodomain, and mature domain), and peptidase property. (b) Structure of cathepsin L. Cys138–His276–Asn300 triad at the active side is colored (model created with SWISS-MODEL and PyMOL). (c) Scheme Cts maturation as the function of endolysosomal pH. The mannose-6-phosphate pathway favors Cts delivery to endosomes, where they are eventually sorted into the lysosomes. At acidic pH, the pro-peptide is removed, and an active single-chain intermediate is generated. The removal of propeptide is mediated by other proteases (independent of autocatalytic activity). The single-chain molecule is further processed into mature two-chain form comprising an amino-terminal light chain and a carboxyl-terminal heavy chain.
Cts B, L, and H are expressed in most cell types, and they are involved in lysosomal nonspecific bulk protein degradation [21], while Cts S, K, V, F, C, and W show more tissue-specific expression and function [5][18][22]. All Cts are monomeric and single domain enzymes, and their structures are composed of two subdomains referred to as the L (left)- and the R (right)-domain, except for CtsC, which is present as a homotetramer (Figure 1b) [23][24]. In its native form, the Cts amino acid sequence includes a signal peptide, a propeptide, and a catalytic domain. The signal peptides can vary between 10 and 20 amino acids and mediate Cts translocation into the endoplasmic reticulum where, after they are cleaved, the inactive precursor is glycosylated [5]. These proteins are further transported to the Golgi apparatus, where the mannose oligosaccharides are phosphorylated, inducing their further translocation into the lysosomes via interaction with the mannose-6-phosphate receptor (M6PR) [25].
The propeptide sequences, which can range from 38 to 251 amino acids (Figure 1a) [21], have a pivotal role in regulating the folding of the catalytic domains, favoring Cts transport in the endo-lysosomal compartment and inhibiting the premature activation of the catalytic domain [26]. Exceptions to this process include CtsH, which is trafficked from the Golgi to the lysosome via the sortilin receptor [27]. The maturation of these zymogens occurs via autocatalytic cleavage at low pH for the endopeptidases (CtsH, L, S, K, V, F, B, and H), whereas the exopeptidases (CtsC and X) are processed by other endopeptidases, such as the CtsS and the CtsL (Figure 1c) [18][20][28] The mature form of Cts contains a disulfide-linked heavy and light chain subunit, and it has an overall molecular weight that ranges from 20 to 35 kDa (except for CtsC that has a molecular weight of 50 kDa). Cts are usually localized in the acidic and reducing environment of the endo-lysosomal vesicles, but their expression and activity have also been detected in the cell nucleus, the plasma membrane, and cytoplasm. For example, CtsW is expressed in the endoplasmic reticulum of natural killer cells, and Cts K was found extracellularly and intracellularly in vesicles, granules, and vacuoles of osteoclasts and chondroclasts. In dendritic cells, Cts F is located in the Golgi apparatus of immature cells, while it is transferred to the endosomal/lysosomal vesicles in mature cells [29][30][31]. The nuclear localization of CtsL and localization of CtsB in plasma membrane correlated with metastatic tumors [32][33][34].
3. Cell Death
The endo/lysosomal compartment was shown to be involved in cell death, and many pieces of evidence support the hypothesis that some Cts could be involved in the regulation of apoptosis [35]. However, Cts positive or negative involvement in the cell death regulation depends on the cellular context, the Cts type [35][36][37][38], and, above all, on their release into the cell cytoplasm [39]. While apoptosis is frequently described as a result of caspase cascade activation, Cts can initiate, during the early phases of cell death, a possible caspase-independent process together with calpains and serine proteases [40][41]. CtsB, H, L, and S play an essential role in cleaving the classic caspase substrates, such as procaspase-1,-3,-7-8 [40][41], and in the release of pro-apoptotic factors such as cytochrome c from mitochondria [42][43], which leads to caspase activation and apoptosis. In this context, Cts were shown to target Bid (BH3 interacting domain death agonist) and/or degrade prosurvival Bcl-2 homologs, thereby triggering the pro-apoptotic activity of Bak (BCL2 antagonist/killer) and Bax (BCL2 associated X, apoptosis regulator) [44][45][46]. Moreover, XIAP (X-chromosome-linked inhibitor of apoptosis) was also found to be a target of Cts, suggesting that they can also mediate mitochondria caspase-dependent apoptosis [38].
The release of the Cts in the cell cytoplasm can occur through different mechanisms/factors including the activation of membrane receptors (such as tumor necrosis factor receptor 1 [43][47]) or the generation of reactive oxygen species (ROS) [48]. It was reported that the ligation of the tumor necrosis factor (TNF) receptor-1 by TNF-α in hepatocytes could cause the activation of caspase-8, which in turn labilizes the lysosomes favoring the release of CtsB. As a result of this phenomenon, mitochondrial release of cytochrome c (activating caspase-9 and eventually caspase-3) occurs, leading to apoptotic cell death [43]. The lysosome destabilization via ROS is regulated by an intraorganelle pool of redox-active iron, which sensitizes lysosomes to oxidative damage [49]. Furthermore, after UVA irradiation, the extracellular lysosomal contents are released and accompanied by translocation of lysosome-associated membrane protein-1 (LAMP-1) to the plasma membrane, which provides evidence for lysosomal exocytosis under stress conditions [50]. While new therapeutic interventions based on lysosomal permeabilization are under investigation to target apoptosis-resistant cancer cells [51], the release of the Cts into the cell cytoplasm can also be a result of chemotherapy [52]. In human hormone-refractory prostate cancer cells (HRPC), docetaxel treatment induced caspase-independent cell death through CtsB activity [53]. A similar mechanism was shown in non-small cell lung cancer (NSCLC) cells, where the microtubules stabilizing agent paclitaxel activates the release of CtsB via disruption of lysosomes in the cell cytoplasm, favoring cell death [54].
4. Autophagy
Autophagy is a lysosomal-dependent, intracellular self-digestion process in which damaged proteins and organelles are transported to the lysosome for degradation to maintain cellular homeostasis. Autophagy prevents cancer onset by removing damaged proteins and organelles, reducing ROS, and promoting the autophagic cell death. During apoptosis, Cts are released from the lysosomes into the cytoplasm, activating degradation cascades, whereas in autophagy, the lysosomes fuse with autophagosomes, forming the autophagolysosomes [55][56]. Alternatively, at very aggressive stages of tumor development, autophagy supports cancer development by protecting the malignant cells from cellular stress, supporting cancer progression [57]. Autophagy is initiated by the generation of a phagophore followed by autophagosomal formation and lysosomal fusion (autolysosome) [58]. Under stress conditions (e.g., hydrogen peroxide or starvation) the number of lysosomes decreases due to their continuous fusion with autophagosomes, which in turn increase [59]. This phenomenon is also responsible for clearing damaged lysosomes after permeabilization [60]. Recently it was shown that there is an active role of the Cts in the molecular switch between apoptosis and autophagy. In MCF-7-breast cancer cells, the treatment with the Cts inhibitor E64d increased autophagosome formation even when used in combination with the apoptosis inducer camptothecin [61]. The inhibition of CtsB and L was shown to favor autophagocytosis over apoptosis, where this process was accompanied by an accumulation of cellular stress and autophagic markers. The un-degraded proteins led to the induction of apoptosis, indicating that the balance between these two phenomena is controlled by many factors [62][63]. In neuroblastoma models, the inhibition of these two Cts resulted in significant accumulation in the cell cytoplasm of large cytoplasmic electron-dense vesicles and generation of multivesicular bodies, while in a double knockout model for CTSB and L, autophagic-like vacuoles accumulated over time [64]. Cts inhibition and consequent autophagocytosis were eventually linked to the dysregulation of the IGF-1 receptor/MAPK/Akt pathway, which is required for tumor cell growth and survival [65]. Interestingly, the inhibition of autophagy using Thymoquinone in glioblastoma cells induced CtsB leakage into the cytosol and caused activation of caspase-independent apoptosis [66], demonstrating that these phenomena are strictly linked to each other.
We can summarize that Cts may be key regulators of pro-survival autophagy in cancer cells, and Cts inhibition could increase the efficacy of cancer treatment focused at inhibiting autophagy. On the other hand, lysosomal destabilization could inhibit the metabolization of substrates generated through autophagy, prevent clearance of ROS, and increase Cts release in the cell cytoplasm where their activity can amplify the apoptotic signaling.
5. Tumor Matrix Cellular Degradation
The EMT is considered an essential process at the base of tumor cell motility and invasiveness. EMT is triggered by multiple transcriptional and biochemical cascades, and it results in the acquisition of migratory and invasive properties typical of mesenchymal cells [67].
In this context, the CtsL is involved in EMT, and its overexpression in lung cancer cells leads—via nuclear factor kappa B (NF-κB) signaling—to the upregulation of nuclear factors belonging to the snail family transcriptional repressors Snail and Slug, and the zinc finger e-box binding homeobox proteins ZEB1 and ZEB2 [68]. Also, Snail can promote the nuclear translocation of CtsL where it degrades its substrate homeobox protein Cux-1, leading to the inhibition of E-cadherin expression and the induction of Snail transcription that promotes EMT [69]. Similar observations were described in cell lines derived from lung and breast cancer (A549 and MCF-7, respectively), where EMT induced by TGF-β activation was associated with the increased protein expression of CtsL and Snail via phosphatidylinositol 3-kinase (PI3K)-AKT and Wnt signaling pathways. Additionally, CTSL knock-down in A549 cells favored mesenchymal to epithelial transition (MET) in vivo, inhibiting xenograft tumor growth [68][70]. EMT regulation by CtsB is connected via the E-box element regulation, which controls EMT activator factors [71]. Besides, it was shown that the Cts B expression knock-down was followed by an increase in Cts X expression compensating the absence of this protease [72][73]. Cts X overexpression was also associated with EMT of hepatocellular cancer and with the upregulation of matrix metalloproteinases (MMP2, MMP3, and MMP9) [74].
In this scenario, Cts cover a central role in tumor progression when they are released in the ECM. They can be secreted or associated with the plasma membrane and caveolae [75]. When secreted, Cts can favor cancer cell spreading by modifying and degrading the ECM and/or activating the matrix metalloproteinases (MMPs) and urokinase plasminogen activator [76][77]. Also, the acidic extracellular environment of the tumor is ideal for favoring the proteolytic activation of the Cts when they are secreted [76][78]. It was demonstrated that ECM degradation by the active form of Cts B and X increased the release of growth factors, including TGFβ-1, an EMT activator [74][79], while regulating cell adhesion.
In the context of the tumor neo-angiogenesis and cell–cell communication, cancer cells can stimulate endothelial cells through two main ways: (1) in a direct manner via secretion of soluble factors and expression of adhesion receptors, gap junctions/tunneling nanotubes, and microvesicles (MVs) [80][81][82]; and (2) in an indirect manner by secretion of proteases into the extracellular space or by changing the pH [78][83][84][85]. One of the causes of resistance to anti-angiogenic therapies has been attributed to the ability of cancer cells to communicate with endothelial cells [84]. Lysosomal exocytosis of Cts such as B, H, and L can regulate this phenomenon in various tissues [7][86][87][88]. For example, the increased secretion of CtsL via lysosome exocytosis was correlated with enhanced tumor cell migration and invasion [7]. HT 1080 cells were shown to secrete CtsL directly into the medium through a lysosome exocytosis independent pathway, and the secreted protein was approximately 10-fold more active (32 kDa) than that detected in the cells [89].
Inhibition of the gene expression of Cts B in glioblastoma cells attenuated their migration and invasion while reducing cell–cell interaction in human microvascular endothelial cells in both in vitro and in vivo models [90].
The acidification of the tumor microenvironment has been shown to increase invasiveness and metastasis, suggesting that it offers a favorable advantage for cancer development, while it is toxic for healthy stromal cells [91]. Also, tumor acidic cell environment increases the bioavailability of many growth factors (e.g., VEGF) by inducing MVs rupture in the microenvironment [92], also favoring the release of CtsB [92], as shown in ovarian, breast, and colon carcinoma three-dimensional (3D) cultures [78][93][94]. The remodeling of the ECM is critical for the tubulogenesis of endothelial cells during cancer vascularization. CtsB was found in human umbilical vein endothelial cells to co-localize within intracellular vesicles and caveolae and participate in cell polarization and migration via secretion of MVs [93]. It was reported that active CtsS controls the production of matrix-derived angiogenic factors such as type IV collagen, canstatin, arresten, and tumstatin [95]. The anti-Cts S antibody Fsn0503 also potentiated the antiangiogenic effects of anti-VEGF therapies [96].
CtsL plays a crucial role in the invasive and the functional capacities of endothelial progenitor cells (EPC) in the EPC-mediated neovascularization process [97]. Gene expression analysis on breast tissues revealed significant upregulation of CTSL and strong correlation with increased relapse and metastatic incidence. Silencing of CTSL using shRNA or KGP94 (a small inhibitor of CtsL) treatment led to a significant reduction in MDA-MB-231 cells to induce angiogenesis in vivo. Moreover, the analyses with KGP94 in vitro demonstrated a considerable decrease in angiogenic properties such as cell sprouting, migration, invasion, tube formation, and proliferation [7]. CtsK and Cts B are essential for extracellular components, such as type I and IV collagens or elastin [98][99], and many studies demonstrated the critical role of CtsK and Cts B in neovascularization [100][101]. The increase of CtsX expression in tumor cells and tumor-associated immune cells is associated with progression and metastasis of gastric cancer, prostate cancer, hepatocellular carcinoma, and malignant melanoma [74][102][103][104]. CtsX modulates cell adhesion and migration by modulating the interaction with integrin receptors, thus supporting invasion through the ECM [105][106].
6. Crosstalk between Cell Death, Autophagy, and Tumor Matrix Degradation
The aforementioned biological processes (cell death, autophagy, and tumor matrix degradation) may synergistically contribute to a cancer drug resistance. While chemotherapeutics were designed to induce apoptosis, their prolonged administration can increase the chance to develop drug resistance phenomena. In this scenario, the induction of autophagic death can be a promising approach to overcome cancer drug resistance. [107]. Autophagy and apoptosis are catabolic pathways essential for homeostasis, and usually they are both considered tumor suppressors. Whereas autophagy is a self-degradative process removing misfolded/aggregated proteins and degraded organelles, apoptosis is the canonical programmed cell death. Several molecular pathways interconnect autophagy and apoptosis, thus any misregulation of these processes can favor cancer cell proliferation. However, autophagy plays a dual role in cancer, as it has recently been shown that this process can also facilitate the survival of tumor cells in stress conditions such as hypoxia or low-nutrition environments [108]. Cts are engaged in all of these biochemical pathways, and they can alternatively favor or inhibit tumor growth. The material destined for degradation enters the lysosomes primarily via endocytosis, autophagy, and phagocytosis and is degraded through the action of hydrolases, including Cts [13]. In autophagy, the functional integrity of the lysosomal compartment provides active degradation, which can counteract apoptosis. In situations when Cts are released into the cytosol upon lysosomal membrane permeabilization, they can amplify the apoptotic signaling or initiate the lysosomal pathway of apoptosis via Bid and/or Bak/Bim cleavage. [109] (Figure 2a,b).
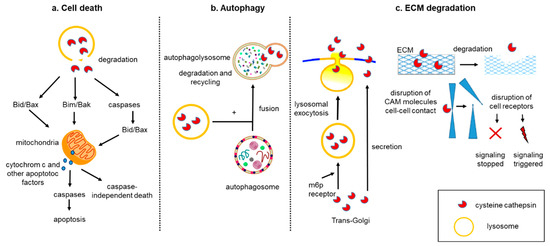
Figure 2. Role of Cts in cell death, autophagy, and ECM degradation. (a) Cts are released into the cytosol and are involved in apoptotic pathways. The first pathway includes a direct cleavage of Bid and/or Bak/Bim, translocation of these pro-apoptotic proteins to the mitochondrial outer membrane, which induces the release of apoptogenic factors such as cytochrome c and subsequent activation of downstream caspases and apoptosis. The second pathway involves a direct cleavage of caspases followed by cleavage of Bid and/or Bax, translocation of these proteins to mitochondria, and similar downstream events leading to apoptosis. The third option is independent-caspase apoptosis. (b) Upon induction of autophagy, cytoplasmic materials are sequestered by a double-membraned structure, autophagosome, which fuses with lysosomes to become autolysosomes. In autolysosome, the sequestered cargos are degraded and recycled. (c) Within the Golgi, Cts are transported into lysosomes via the mannose-6-phosphate (m6p receptor) pathway, but a minor population of Cts (~5%) is not converted to the m6p form and as a result is shunted into the exocytosis pathway [5]. Additionally, this direct secretion pathway of CtsL was detected in human fibrosarcoma cells (HT 1080) [89]. In the extracellular space, Cts cleave different targets. e.g., cell adhesion molecules (CAM), cell–cell contacts, and proteins of ECM, which affect cell adhesion and migration. Proteolytic products of these cleavages can act as signaling molecules and influence growth and invasion of cancer. Next, the cleave of receptors by Cts can result in either constantly triggered or inhibited signalings.
The malignant phenotype of cancer cells is accompanied by ECM degradation and morphological changes, which are typical events characterizing EMT. As a result of this transformation, cells modulate the profile of adhesion proteins, cell receptors, cytoskeleton polarization, and secretion of molecules, such as cytokines, growth factors, and proteases (Figure 2c) [110]. All these events can upregulate cancer motility and invasion properties of tumor cells. Overexpression of primary factors such as Twist, Zeb, Slug, and the activation of signaling pathways such as Hedgehog and TGFβ/Smad4 is induced during EMT and correlates with the onset of drug resistance phenomena [111]. Furthermore, the metastatic spreading relies on newly formed vessels, which are accompanied by proteolytic degradation of the endothelial basement and ECM [112].
7. Drug Resistance
Cancer drug resistance can have many different origins, such as drug inactivation, increased or decreased expression of efflux and influx pumps, respectively, modulation of specific mechanisms of cell death, autophagy, the occurrence of EMT, aberrant cell–cell communication, and epigenetic alterations, amongst others. These phenomena can occur individually or concurrently while affecting the efficacy of a wide range of cancer therapeutics [107]. One of these phenomena is strongly connected to the biology of the lysosomes, since many of the classical chemotherapeutics are sequestered in the acidic pH lumen of these organelles [113], reducing their ability to interact with their targets. It was shown that the amount of drug accumulating in the lysosomes is directly proportional to the cellular tolerance against the cytotoxic effects of the therapeutic [114][115]. Weakly basic, hydrophobic anticancer drugs after internalization may undergo rapid incorporation into lysosomes. Upon entrapment in lysosomes, lysosomotropic compounds appear to inflict various deleterious effects on the membrane of the lysosomes, provoking lysosomal recycling, biogenesis, and exocytosis. As a result of this phenomenon, the lysosomal sequestration of chemotherapeutic drugs in lysosomes prevents them from reaching their intracellular target sites [113]. This was observed for several classical chemotherapeutics, including Doxorubicin [116], Mitoxantrone [117], Sunitinib [118] and Daunorubicin [119], where their internalization was followed by their accumulation in the lysosomes [120].
Another mechanism of drug resistance is related to lysosomal exocytosis, a process in which lysosomes are recruited to the cell surface and fuse with the plasma membrane via a Ca2+-dependent process releasing their cargo (and the entrapped drug) into the extracellular milieu [121]. This phenomenon occurs as an adaptation of cells to the hypoxic and the acidic environment of the tumor. Hypoxia leads to the acidification of the tumor microenvironment, which can trigger proteases secretion, including Cts, from cancer cells to the extracellular space, enhancing the tumor invasiveness [122]. Interestingly, it was shown that oral administration of buffer, which neutralized intratumoral pH, reduced metastasis of cancer cells that was accompanied by lower CtsB activity [123].
The specific contribution of Cts in the occurrence of chemoresistance phenomena is currently under debate. The overexpression of CtsB and L was associated with increased drug inactivation and, above all, with increased lysosome trafficking to the plasma membrane and secretion of lysosomal cargo [124]. As a result of this phenomenon, CTSL knock-down in ovarian cancer cells SCOV3 resulted in increased apoptosis induced by paclitaxel (the most common drug in the management of ovarian cancer) [16]. Also, Cts L played a role in inhibiting the cell senescence process in different tumor cell lines, providing an additional mechanism of protease-mediated drug target elimination (trapping a drug in lysosomes) and drug resistance [15][68][125]. Treatment of drug-resistant neuroblastoma cells with doxorubicin and pan-caspase inhibitor (Q-VD-OPH) significantly decreased cell viability and senescence phenotype, as evidenced by an increased p21/WAF1 expression, senescence-associated β-galactosidase activity, and cell growth arrest. Experiments have established that this phenomenon was the result of CtsL inhibition [15].
In this context, Cts L inhibition increased Doxorubicin cell accumulation and a more favorable nuclear distribution of the drug in the cells despite P-gp expression [125]. The CstL inhibition stabilized and enhanced the availability of cytoplasmic and nuclear drug targets such as estrogen receptor-α, Bcr-Abl, topoisomerase-IIα, histone deacetylase 1, and the androgen receptor I, increasing the cellular response for different therapeutics drugs (doxorubicin, tamoxifen, imatinib, trichostatin A, and flutamide) [125].
These findings provide evidence for the potential role of Cts as targets, and their inhibitors can represent a tool to suppress cancer resistance to chemotherapy and increase their sensitivity to the drugs.
References
- Brix, K. Lysosomal Proteases: Revival of the Sleeping Beauty. In Madame Curie Bioscience Database [Internet]; Paul Saftig; Landes Bioscience: Austin, TX, USA, 2005.
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339.
- Fonović, M.; Turk, B. Cysteine cathepsins and extracellular matrix degradation. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2560–2570.
- Kukor, Z.; Mayerle, J.; Kruger, B.; Toth, M.; Steed, P.M.; Halangk, W.; Lerch, M.M.; Sahin-Toth, M. Presence of cathepsin B in the human pancreatic secretory pathway and its role in trypsinogen activation during hereditary pancreatitis. J. Biol. Chem. 2002, 277, 21389–21396.
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431.
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64.
- Sudhan, D.R.; Siemann, D.W. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin. Exp. Metastasis 2013, 30, 891–902.
- Chen, S.; Dong, H.; Yang, S.; Guo, H. Cathepsins in digestive cancers. Oncotarget 2017, 8, 41690.
- Sever, S.; Altintas, M.M.; Nankoe, S.R.; Möller, C.C.; Ko, D.; Wei, C.; Henderson, J.; del Re, E.C.; Hsing, L.; Erickson, A. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J. Clin. Investig. 2007, 117, 2095–2104.
- Goulet, B.; Baruch, A.; Moon, N.-S.; Poirier, M.; Sansregret, L.L.; Erickson, A.; Bogyo, M.; Nepveu, A. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol. Cell 2004, 14, 207–219.
- Cheng, X.W.; Shi, G.-P.; Kuzuya, M.; Sasaki, T.; Okumura, K.; Murohara, T. Role for cysteine protease cathepsins in heart disease: Focus on biology and mechanisms with clinical implication. Circulation 2012, 125, 1551–1562.
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta. 2000, 291, 113–135.
- Vidak, E.; Javorsek, U.; Vizovisek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264.
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712.
- Zheng, X.; Chou, P.M.; Mirkin, B.L.; Rebbaa, A. Senescence-initiated reversal of drug resistance: Specific role of cathepsin L. Cancer Res. 2004, 64, 1773–1780.
- Sui, H.; Shi, C.; Yan, Z.; Wu, M. Overexpression of Cathepsin L is associated with chemoresistance and invasion of epithelial ovarian cancer. Oncotarget 2016, 7, 45995.
- Turk, V.; Turk, B.; Turk, D. Lysosomal cysteine proteases: Facts and opportunities. EMBO J. 2001, 20, 4629–4633.
- Brömme, D.; Wilson, S. Role of cysteine cathepsins in extracellular proteolysis; Springer: Berlin/Heidelberg, Germany, 2011; pp. 23–51.
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: Essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des. 2002, 8, 1623–1637.
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine proteases: Modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 2016, 7, 107.
- Lecaille, F.; Kaleta, J.; Brömme, D. Human and parasitic papain-like cysteine proteases: Their role in physiology and pathology and recent developments in inhibitor design. Chem. Rev. 2002, 102, 4459–4488.
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207.
- Grzonka, Z.; Jankowska, E.; Kasprzykowski, F.; Kasprzykowska, R.; Lankiewicz, L.; Wiczk, W.; Wieczerzak, E.; Ciarkowski, J.; Drabik, P.; Janowski, R.; et al. Structural studies of cysteine proteases and their inhibitors. Acta Biochim. Pol. 2001, 48, 1–20.
- Stoka, V.; Turk, B.; Turk, V. Lysosomal cysteine proteases: Structural features and their role in apoptosis. IUBMB Life 2005, 57, 347–353.
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 605–614.
- Wiederanders, B. The function of propeptide domains of cysteine proteinases; Springer: Berlin/Heidelberg, Germany, 2002; pp. 261–270.
- Canuel, M.; Korkidakis, A.; Konnyu, K.; Morales, C.R. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem. Biophys. Res. Commun. 2008, 373, 292–297.
- Dahl, S.W.; Halkier, T.; Lauritzen, C.; Dolenc, I.; Pedersen, J.; Turk, V.; Turk, B. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 2001, 40, 1671–1678.
- Yamaza, T.; Goto, T.; Kamiya, T.; Kobayashi, Y.; Sakai, H.; Tanaka, T. Study of immunoelectron microscopic localization of cathepsin K in osteoclasts and other bone cells in the mouse femur. Bone 1998, 23, 499–509.
- Perišić Nanut, M.; Sabotič, J.; Jewett, A.; Kos, J. Cysteine cathepsins as regulators of the cytotoxicity of NK and T cells. Front. Immunol. 2014, 5, 616.
- Magister, Š.; Obermajer, N.; Mirković, B.; Švajger, U.; Renko, M.; Softić, A.; Colbert, J.D.; Watts, C.; Kos, J. Regulation of cathepsins S and L by cystatin F during maturation of dendritic cells. Eur. J. Cell Biol. 2012, 91, 391–401.
- Wendt, W.; Schulten, R.; Stichel, C.C.; Lübbert, H. Intra-versus extracellular effects of microglia-derived cysteine proteases in a conditioned medium transfer model. J. Neurochem. 2009, 110, 1931–1941.
- Sullivan, S.; Tosetto, M.; Kevans, D.; Coss, A.; Wang, L.; O’Donoghue, D.; Hyland, J.; Sheahan, K.; Mulcahy, H.; O’Sullivan, J. Localization of nuclear cathepsin L and its association with disease progression and poor outcome in colorectal cancer. Int. J. Cancer 2009, 125, 54–61.
- Sloane, B.F.; Rozhin, J.; Johnson, K.; Taylor, H.; Crissman, J.D.; Honn, K.V. Cathepsin B: Association with plasma membrane in metastatic tumors. Proc. Natl. Acad. Sci. 1986, 83, 2483–2487.
- Chwieralski, C.; Welte, T.; Bühling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149.
- Boya, P.; Andreau, K.; Poncet, D.; Zamzami, N.; Perfettini, J.-L.; Metivier, D.; Ojcius, D.M.; Jäättelä, M.; Kroemer, G. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J. Exp. Med. 2003, 197, 1323–1334.
- Johansson, A.-C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Öllinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540.
- Droga-Mazovec, G.; Bojič, L.; Petelin, A.; Ivanova, S.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150.
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 22–33.
- Johnson, D. Noncaspase proteases in apoptosis. Leukemia 2000, 14, 1695.
- Leist, M.; Jäättelä, M. Triggering of apoptosis by cathepsins. Cell Death Differ. 2001, 8, 324–326.
- Zhao, M.; Antunes, F.; Eaton, J.W.; Brunk, U.T. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur. J. Biochem. 2003, 270, 3778–3786.
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-α–mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137.
- Cirman, T.; Orešić, K.; Mazovec, G.D.; Turk, V.; Reed, J.C.; Myers, R.M.; Salvesen, G.S.; Turk, B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 2004, 279, 3578–3587.
- Stoka, V.; Turk, B.; Schendel, S.L.; Kim, T.-H.; Cirman, T.; Snipas, S.J.; Ellerby, L.M.; Bredesen, D.; Freeze, H.; Abrahamson, M. Lysosomal protease pathways to apoptosis cleavage of Bid, not pro-caspases, is the most likely route. J. Biol. Chem. 2001, 276, 3149–3157.
- Billen, L.; Shamas-Din, A.; Andrews, D. Bid: A Bax-like BH3 protein. Oncogene 2009, 27, S93.
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 746–754.
- Zdolsek, J.; Zhang, H.; Roberg, K.; Brunk, U.; Sies, H. H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic. Res. Commun. 1993, 18, 71–85.
- Yu, Z.; Persson, H.L.; Eaton, J.W.; Brunk, U.T. Intralysosomal iron: A major determinant of oxidant-induced cell death. Free Radic. Biol. Med. 2003, 34, 1243–1252.
- Appelqvist, H.; Waster, P.; Eriksson, I.; Rosdahl, I.; Ollinger, K. Lysosomal exocytosis and caspase-8-mediated apoptosis in UVA-irradiated keratinocytes. J. Cell Sci. 2013, 126, 5578–5584.
- Groth-Pedersen, L.; Jäättelä, M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013, 332, 265–274.
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64.
- Mediavilla-Varela, M.; Pacheco, F.J.; Almaguel, F.; Perez, J.; Sahakian, E.; Daniels, T.R.; Leoh, L.S.; Padilla, A.; Wall, N.R.; Lilly, M.B. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol. Cancer 2009, 8, 68.
- Bröker, L.E.; Huisman, C.; Span, S.W.; Rodriguez, J.A.; Kruyt, F.A.; Giaccone, G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004, 64, 27–30.
- Kim, J.; Klionsky, D.J. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu. Rev. Biochem. 2000, 69, 303–342.
- Dielschneider, R.F.; Henson, E.S.; Gibson, S.B. Lysosomes as oxidative targets for cancer therapy. Oxidative Med. Cell. Longev. 2017, 2017.
- Liu, B.; Wen, X.; Cheng, Y. Survival or death: Disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013, 4, e892.
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12.
- Zhang, J.; Zhou, W.; Lin, J.; Wei, P.; Zhang, Y.; Jin, P.; Chen, M.; Man, N.; Wen, L. Autophagic lysosomal reformation depends on mTOR reactivation in H2O2-induced autophagy. Int. J. Biochem. Cell Biol. 2016, 70, 76–81.
- Hasegawa, J.; Maejima, I.; Iwamoto, R.; Yoshimori, T. Selective autophagy: Lysophagy. Methods 2015, 75, 128–132.
- Lamparska-Przybysz, M.; Gajkowska, B.; Motyl, T. Cathepsins and BID are involved in the molecular switch between apoptosis and autophagy in breast cancer MCF-7 cells exposed to camptothecin. J. Physiol. Pharmacol. 2005, 56, 159.
- Cartledge, D.M.; Colella, R.; Glazewski, L.; Lu, G.; Mason, R.W. Inhibitors of cathepsins B and L induce autophagy and cell death in neuroblastoma cells. Investig. New Drugs 2013, 31, 20–29.
- Colella, R.; Lu, G.; Glazewski, L.; Korant, B.; Matlapudi, A.; England, M.R.; Craft, C.; Frantz, C.N.; Mason, R.W. Induction of cell death in neuroblastoma by inhibition of cathepsins B and L. Cancer Lett. 2010, 294, 195–203.
- Stahl, S.; Reinders, Y.; Asan, E.; Mothes, W.; Conzelmann, E.; Sickmann, A.; Felbor, U. Proteomic analysis of cathepsin B and L-deficient mouse brain lysosomes. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2007, 1774, 1237–1246.
- Soori, M.; Lu, G.; Mason, R.W. Cathepsin Inhibition Prevents Autophagic Protein Turnover and Downregulates Insulin Growth Factor-1 Receptor–Mediated Signaling in Neuroblastoma. J. Pharmacol. Exp. Ther. 2016, 356, 375–386.
- Racoma, I.O.; Meisen, W.H.; Wang, Q.-E.; Kaur, B.; Wani, A.A. Thymoquinone inhibits autophagy and induces cathepsin-mediated, caspase-independent cell death in glioblastoma cells. PLoS ONE 2013, 8, e72882.
- Gos12, M.; Joanna Miłoszewska, M.P. Epithelial-mesenchymal transition in cancer progression. POSTĘPY Biochem. 2009, 55, 121–128.
- Han, M.-l.; Zhao, Y.-f.; Tan, C.-h.; Xiong, Y.-j.; Wang, W.-j.; Wu, F.; Fei, Y.; Wang, L.; Liang, Z.-q. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol. Sin. 2016, 37, 1606.
- Fei, Y.; Xiong, Y.; Shen, X.; Zhao, Y.; Zhu, Y.; Wang, L.; Liang, Z. Cathepsin L promotes ionizing radiation-induced U251 glioma cell migration and invasion through regulating the GSK-3β/CUX1 pathway. Cell. Signal. 2018, 44, 62–71.
- Zhang, Q.; Han, M.; Wang, W.; Song, Y.; Chen, G.; Wang, Z.; Liang, Z. Downregulation of cathepsin L suppresses cancer invasion and migration by inhibiting transforming growth factor-β-mediated epithelial-mesenchymal transition. Oncol. Rep. 2015, 33, 1851–1859.
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291.
- Lechner, A.M.; Assfalg-Machleidt, I.; Zahler, S.; Stoeckelhuber, M.; Machleidt, W.; Jochum, M.; Nägler, D.K. RGD-dependent binding of procathepsin X to integrin αvβ3 mediates cell-adhesive properties. J. Biol. Chem. 2006, 281, 39588–39597.
- Sevenich, L.; Schurigt, U.; Sachse, K.; Gajda, M.; Werner, F.; Müller, S.; Vasiljeva, O.; Schwinde, A.; Klemm, N.; Deussing, J. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 2497–2502.
- Wang, J.; Chen, L.; Li, Y.; Guan, X.-Y. Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS ONE 2011, 6, e24967.
- Cavallo-Medved, D.; Mai, J.; Dosescu, J.; Sameni, M.; Sloane, B.F. Caveolin-1 mediates the expression and localization of cathepsin B, pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J. Cell Sci. 2005, 118, 1493–1503.
- Affara, N.I.; Andreu, P.; Coussens, L.M. Delineating protease functions during cancer development; Proteases and Cancer, Humana Press: New York, NY, USA, 2009; pp. 1–32.
- Kirschke, H.; Eerola, R.; Hopsu-Havu, V.; Brömme, D.; Vuorio, E. Antisense RNA inhibition of cathepsin L expression reduces tumorigenicity of malignant cells. Eur. J. Cancer 2000, 36, 787–795.
- Rothberg, J.M.; Bailey, K.M.; Wojtkowiak, J.W.; Ben-Nun, Y.; Bogyo, M.; Weber, E.; Moin, K.; Blum, G.; Mattingly, R.R.; Gillies, R.J. Acid-mediated tumor proteolysis: Contribution of cysteine cathepsins. Neoplasia 2013, 15, 1125–IN1129.
- Kasabova, M.; Joulin-Giet, A.; Lecaille, F.; Gilmore, B.F.; Marchand-Adam, S.; Saidi, A.; Lalmanach, G. Regulation of TGF-β1-driven differentiation of human lung fibroblasts: Emerging roles of cathepsin B and cystatin C. J. Biol. Chem. 2014, 289, 16239–16251.
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569.
- Munson, P.; Shukla, A. Exosomes: Potential in cancer diagnosis and therapy. Medicines 2015, 2, 310–327.
- Van Hinsbergh, V.W.; Engelse, M.A.; Quax, P.H. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 716–728.
- Rath, B.; Klameth, L.; Plangger, A.; Hochmair, M.; Ulsperger, E.; Huk, I.; Zeillinger, R.; Hamilton, G. Expression of Proteolytic Enzymes by Small Cell Lung Cancer Circulating Tumor Cell Lines. Cancers 2019, 11, 114.
- Lopes-Bastos, B.M.; Jiang, W.G.; Cai, J. Tumour–Endothelial Cell Communications: Important and Indispensable Mediators of Tumour Angiogenesis. Anticancer Res. 2016, 36, 1119–1126.
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535.
- Uchiyama, Y.; Waguri, S.; Sato, N.; Watanabe, T.; Ishido, K.; Kominami, E. Cell and tissue distribution of lysosomal cysteine proteinases, cathepsins B, H, and L, and their biological roles. Acta Histochem. Et Cytochem. 1994, 27, 287–308.
- Gocheva, V.; Chen, X.; Peters, C.; Reinheckel, T.; Joyce, J.A. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol. Chem. 2010, 391, 937–945.
- Liu, Y.; Zhou, Y.; Zhu, K. Inhibition of glioma cell lysosome exocytosis inhibits glioma invasion. PLoS ONE 2012, 7, e45910.
- Hashimoto, Y.; Kondo, C.; Kojima, T.; Nagata, H.; Moriyama, A.; Hayakawa, T.; Katunuma, N. Significance of 32-kDa cathepsin L secreted from cancer cells. Cancer Biother. Radiopharm. 2006, 21, 217–224.
- Lakka, S.S.; Gondi, C.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 2004, 23, 4681.
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223.
- Giusti, I.; D’Ascenzo, S.; Millimaggi, D.; Taraboletti, G.; Carta, G.; Franceschini, N.; Pavan, A.; Dolo, V. Cathepsin B mediates the pH-dependent proinvasive activity of tumor-shed microvesicles. Neoplasia 2008, 10, 481–488.
- Cavallo-Medved, D.; Rudy, D.; Blum, G.; Bogyo, M.; Caglic, D.; Sloane, B.F. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp. Cell Res. 2009, 315, 1234–1246.
- Caglič, D.; Pungerčar, J.R.; Pejler, G.; Turk, V.; Turk, B. Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 2007, 282, 33076–33085.
- Wang, B.; Sun, J.; Kitamoto, S.; Yang, M.; Grubb, A.; Chapman, H.A.; Kalluri, R.; Shi, G.-P. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J. Biol. Chem. 2006, 281, 6020–6029.
- Ward, C.; Kuehn, D.; Burden, R.E.; Gormley, J.A.; Jaquin, T.J.; Gazdoiu, M.; Small, D.; Bicknell, R.; Johnston, J.A.; Scott, C.J. Antibody targeting of cathepsin S inhibits angiogenesis and synergistically enhances anti-VEGF. PLoS ONE 2010, 5, e12543.
- Urbich, C.; Heeschen, C.; Aicher, A.; Sasaki, K.-i.; Bruhl, T.; Farhadi, M.R.; Vajkoczy, P.; Hofmann, W.K.; Peters, C.; Pennacchio, L.A. Cathepsin L is required for endothelial progenitor cell–induced neovascularization. Nat. Med. 2005, 11, 206.
- Kusunoki, T.; Nishida, S.; Nakano, T.; Funasaka, K.; Kimoto, S.; Murata, K.; Tomura, T. Study on cathepsin B activity in human thyroid tumors. Auris Nasus Larynx 1995, 22, 43–48.
- Novinec, M.; Grass, R.N.; Stark, W.J.; Turk, V.; Baici, A.; Lenarčič, B. Interaction between Human Cathepsins K, L, and S and Elastins mechanism of elastinolysis and inhibition by macromolecular inhibitors. J. Biol. Chem. 2007, 282, 7893–7902.
- Zhou, G.; Yu, W.; Li, X. The experimental study of specific inhibitor-CA-074Me of Cathepsin B suppressing retinal neovascularization. [Zhonghua Yan Ke Za Zhi] Chin. J. Ophthalmol. 2008, 44, 207–211.
- Jiang, H.; Cheng, X.W.; Shi, G.-P.; Hu, L.; Inoue, A.; Yamamura, Y.; Wu, H.; Takeshita, K.; Li, X.; Huang, Z. Cathepsin K-mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat. Commun. 2014, 5, 3838.
- Krueger, S.; Kalinski, T.; Hundertmark, T.; Wex, T.; Küster, D.; Peitz, U.; Ebert, M.; Nägler, D.K.; Kellner, U.; Malfertheiner, P. Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2005, 207, 32–42.
- Nägler, D.K.; Krüger, S.; Kellner, A.; Ziomek, E.; Menard, R.; Buhtz, P.; Krams, M.; Roessner, A.; Kellner, U. Up-regulation of cathepsin X in prostate cancer and prostatic intraepithelial neoplasia. Prostate 2004, 60, 109–119.
- Rumpler, G.; Becker, B.; Hafner, C.; McClelland, M.; Stolz, W.; Landthaler, M.; Schmitt, R.; Bosserhoff, A.; Vogt, T. Identification of differentially expressed genes in models of melanoma progression by cDNA array analysis: SPARC, MIF and a novel cathepsin protease characterize aggressive phenotypes. Exp. Dermatol. 2003, 12, 761–771.
- Jechorek, D.; Votapek, J.; Meyer, F.; Kandulski, A.; Roessner, A.; Franke, S. Characterization of cathepsin X in colorectal cancer development and progression. Pathol. Res. Pract. 2014, 210, 822–829.
- Obermajer, N.; Premzl, A.; Bergant, T.Z.; Turk, B.; Kos, J. Carboxypeptidase cathepsin X mediates β2-integrin-dependent adhesion of differentiated U-937 cells. Exp. Cell Res. 2006, 312, 2515–2527.
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792.
- Yonekawa, T.; Thorburn, A. Autophagy and Cell Death. Essays Biochem. 2013, 55, 105–117.
- Conus, S.; Simon, H.U. Cathepsins: Key modulators of cell death and inflammatory responses. Biochem. Pharm. 2008, 76, 1374–1382.
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Nat. 2015, 7, 17–28.
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8.
- Gordon, M.S.; Mendelson, D.S.; Kato, G. Tumor angiogenesis and novel antiangiogenic strategies. Int. J. Cancer 2010, 126, 1777–1787.
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117.
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143.
- Guo, B.; Tam, A.; Santi, S.A.; Parissenti, A.M. Role of autophagy and lysosomal drug sequestration in acquired resistance to doxorubicin in MCF-7 cells. BMC Cancer 2016, 16, 762.
- Schindler, M.; Grabski, S.; Hoff, E.; Simon, S.M. Defective pH regulation of acidic compartments in human breast cancer cells (MCF-7) is normalized in adriamycin-resistant cells (MCF-7adr). Biochemistry 1996, 35, 2811–2817.
- Smith, P.J.; Sykes, H.R.; Fox, M.E.; Furlong, I.J. Subcellular distribution of the anticancer drug mitoxantrone in human and drug-resistant murine cells analyzed by flow cytometry and confocal microscopy and its relationship to the induction of DNA damage. Cancer Res. 1992, 52, 4000–4008.
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; De Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346.
- Gong, Y.; Duvvuri, M.; Duncan, M.B.; Liu, J.; Krise, J.P. Niemann-Pick C1 protein facilitates the efflux of the anticancer drug daunorubicin from cells according to a novel vesicle-mediated pathway. J. Pharmacol. Exp. Ther. 2006, 316, 242–247.
- Regev, R.; Yeheskely-Hayon, D.; Katzir, H.; Eytan, G.D. Transport of anthracyclines and mitoxantrone across membranes by a flip-flop mechanism. Biochem. Pharmacol. 2005, 70, 161–169.
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321.
- Asgharzadeh, M.R.; Barar, J.; Pourseif, M.M.; Eskandani, M.; Niya, M.J.; Mashayekhi, M.R.; Omidi, Y. Molecular machineries of pH dysregulation in tumor microenvironment: Potential targets for cancer therapy. BioImpacts BI 2017, 7, 115.
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268.
- Hämälistö, S.; Jäättelä, M. Lysosomes in cancer—Living on the edge (of the cell). Curr. Opin. Cell Biol. 2016, 39, 69–76.
- Zheng, X.; Chu, F.; Chou, P.M.; Gallati, C.; Dier, E.; Mirkin, B.L.; Mousa, S.A.; Rebbaa, A. Cathepsin L inhibition suppresses drug resistance in vitro and in vivo: A putative mechanism. Am. J. Physiol. Cell Physiol. 2009, 296, C65–C74.


