 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vincenzo Piccolo | + 1337 word(s) | 1337 | 2021-02-01 10:05:19 | | | |
| 2 | Vivi Li | + 1 word(s) | 1338 | 2021-02-02 03:24:21 | | |
Video Upload Options
Neurofibromatosis type 1 (NF1) is a complex autosomal dominant disorder associated with germline mutations in the NF1 tumor suppressor gene. NF1 belongs to a class of congenital anomaly syndromes called RASopathies, a group of rare genetic conditions caused by mutations in the Ras/mitogen-activated protein kinase pathway. Generally, NF1 patients present with dermatologic manifestations.
1. Background
Neurofibromatosis type 1 (NF1) is a complex autosomal dominant disorder associated with germline mutations in the NF1 tumor suppressor gene [1]. NF1 belongs to a class of congenital anomaly syndromes called RASopathies, a group of rare genetic conditions caused by mutations in the Ras/mitogen-activated protein kinase (RAS/MAPK) pathway [2]. Generally, NF1 patients present with dermatologic manifestations (Table 1). Other abnormalities in the nervous system and skeletal system may also develop [3].
Table 1. Summary of main features of cutaneous findings of Neurofibromatosis type 1 (NF1).
| Cafe-au-lait Macules | Light to dark brown-pigmented, oval or round skin patches with regular, well-demarcated margin |
| Freckles | Small, clustered pigmented lentigines |
| Neurofibroma | Dome—shaped, pink to brown, pedunculated or sessile cutaneous tumors |
| Plexiform Neurofibroma | Thickened, slightly raised and pigmeted firm masses located along the nerve; “Bag of worms” appearance; hypertrichosis |
| Nevus Anemicus | Hypopigmented, pale, well-defined skin patch |
| Juvenile Xantogranuloma | Yellowish-pink dome-shaped granulomatous papules and nodules |
| Glomus Tumor | Small reddish-blue papulonodule often located at the subungual area |
| Melanoma | Unevenly pigmented unusual looking macule |
2. Cafe-Au-Lait Macules
A CALM is a pigmented, flat skin patch which is the most common initial manifestation of NF-1. CALM is seen in 99% of NF1 patients. The classical CALM lesion in NF1 has a round or oval shape with a uniform color of light to dark brown and regular well-demarcated margins (“coast of California”) [4]. CALMs may be observed at birth, become almost always obvious by the first year of life, and may increase in diameter and number. NF1 patients have higher numbers of CALMs widely distributed on any part of the body except for palms and soles. CALMs rarely occur on the face. Although one to three macules may be present in up to 15% of the normal population, six or more of these lesions greater than 0.5 cm in prepubertal children or greater than 1.5 cm after puberty are diagnostics for NF1 [5]. However, it is also recommended to evaluate and follow the patients with more than three CALMs, as it is less frequent to see more than three lesions in the normal population [6]. There is no relation between NF1 severity and number of lesions [7]. Macules may get tanned under UV exposure and Wood′s lamp may be helpful to detect CALMs invisible to the naked eye [8]. Underlying PNF may accompany CALMs, especially those with hypertrichosis. Therefore, palpation is recommended [9] (Figure 1, Figure 2, Figure 3 and Figure 4).
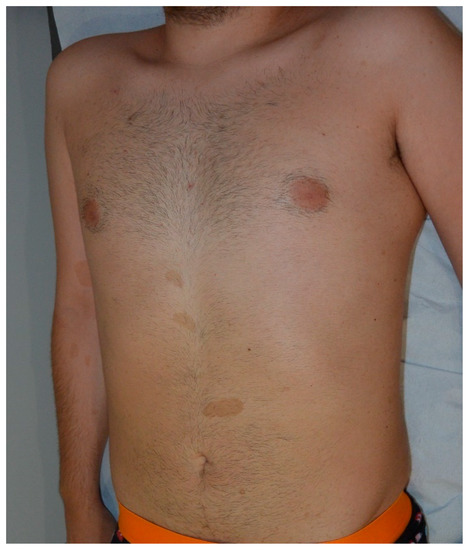
Figure 1. Multiple typical cafe-au-lait macules (CALMs) on the trunk of a young man.
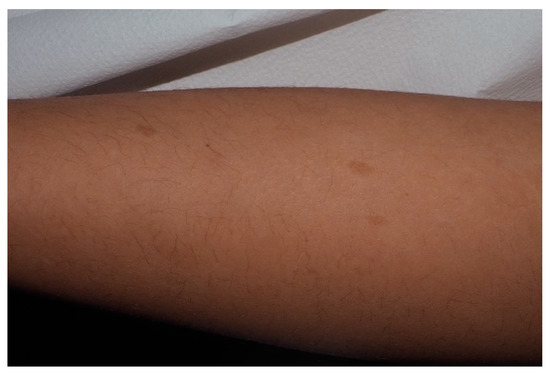
Figure 2. For this child, CALMs were detectable on lower limbs. Note that the size is larger than 5 mm and the typical well-demarcated borders.
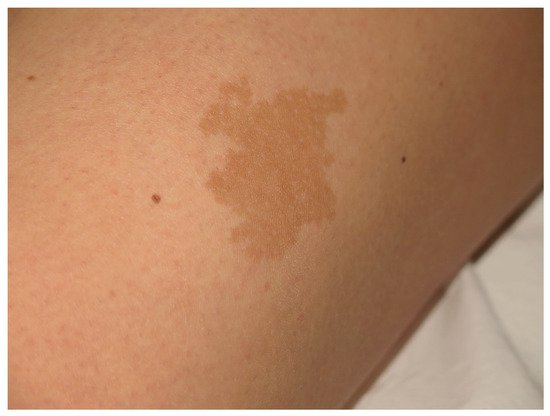
Figure 3. This CALM is not associated with NF1. It shows irregular borders and it is isolated.
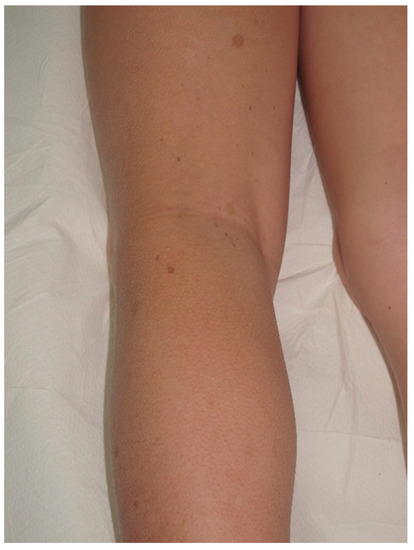
Figure 4. Small CALMs of the left thigh of a young child associated with early flexural freckling.
Another type of CALM which has irregular jagged margins (“coast of Maine”) is mostly solitary and larger; arrangement in a blaschkoid pattern is suggestive of sporadic cases or other genetic disorders. Additionally, it is extraordinary for new sporadic CALMs to occur after the age of 6 [4].
Histological findings are basal hyperpigmentation and sometimes giant melanosomes (macromelanasomes), although diagnosis is clinical and pathology rarely necessary. Recently, mutation of both alleles of the NF1 gene has been detected in the melanocytes of NF1-CALM while normal skin has a germline mutation [10]. CALMs in NF1 tend to show greater melanocyte density and greater stem-cell factor secretion (KIT ligand) of fibroblasts while non-NF1 CALM lesions show the same melanocyte level compared to the normal skin. A melanocyte activation response to hepatocyte growth factor and KIT ligand has been hypothesized as the mechanism of melanocyte growth and mast cell infiltration [11][12].
3. Malignant Peripheral Nerve Sheath Tumors
MPNSTs are aggressive sarcomas which may develop in 3–15% of patients with NF1 [13]. They mostly transform from plexiform or nodular neurofibromas predominantly after puberty. Especially in NF1 affected young adults, the rapid expansion of the tumor with pain or the presence of new neurological symptoms should alert the clinician for MPNST [14]. Coffien et al. reported NF1 patients with MPNST occurring as a second malignant neoplasm especially after alkylating agent chemotherapy and irradiation [15]. Positron emission tomography imaging is shown to be a more useful diagnostic tool to reveal these malignant degenerations [16].
Although the inactivation of the loss of biallelicity of the NF1 gene in Schwann cells seems to be an important initiating factor in the development of MPNST, genomic studies show that the malignant degeneration arises as a result of some additional alterations. Somatic loss-of-function mutations or deletions of the Polycomb repressive complex 2 (PRC2) its core subunits (EED or SUZ12) have been described in NF-1-associated MPNSTs. PRC2 plays a role in chromatin compaction, cell proliferation, differentiation and stem-cell plasticity. A cooperative role of TP53, epidermal growth factor receptor (EGFR) inactivation has also been proposed to contribute MNPST transformation. CDKN2A loss has also been linked to early steps of MPNST [17].
While high-grade MPNST often metastasizes to bone and lung, low-grade tumors have a better prognosis. The main therapy remains a complete surgical excision with negative margins [18]. Discussions on the benefits of adjuvant/neoadjuvant chemotherapies continue. Epirubicin-based adjuvant/neoadjuvant chemotherapy appears to show some survival benefit [19]. Although the development of radiotherapy-induced neoplasms is known to be increased in NF1 patients, radiotherapy may be useful for downsizing the tumors. On the other hand, research shows that radiotherapy benefits local control of the tumor without contributing to survival [20].
Metastatic disease remains challenging due to a lack of promising therapies. Chemotherapy is known to be the standard approach. Biological therapies combining MEK/mTOR inhibition are still under investigation in preclinical and clinical trials. Pexidartinib (tyrosine kinase inhibitor) and sirolimus (mTOR inhibitor) combination contributed to stabilize the disease in a phase I study [21]. Additionally, immunotherapy has also opened up a method for the treatment of MPNST [22].
In terms of the future direction of the treatments, gene-based therapies targeting PRC2, the Janus Kinase (JAK) pathway and HIPPO pathway are presented as a new hope. EZH2 inhibitors—such as tazemetostat for the PRC2 complex, Tyrosine Kinase 2 (TYK2) inhibitors targeting the JAK/STAT pathway and verteporfin inhibiting the HIPPO pathway—could be considered in the combination treatments [23].
4. Optic Pathway Glioma
NF1 is one of the most common heritable brain tumor predisposition disorders, in which affected individuals develop low-grade gliomas classified as WHO grade I gliomas (pilocytic astrocytomas). They are generally indolent neoplasms usually occurring before the age of 8 located within the optic pathway [24]. OPGs occur in 15–20% of NF1 patients [25]. They are often asymptomatic until the age of 7. The tumor may arise from anywhere along the optic pathway such as nerves, chiasm, postchiasmatic tracts, and radiations. Tumor location strongly affects the symptom spectrum. While 30–50% of OPGs become symptomatic, patients may present with unilateral proptosis, visual acuity loss, visual field defect, strabismus, relative afferent pupillary defect, and optic papilloedema or optic nerve atrophy. Chiasmal optic pathway gliomas can present with precocious puberty without any visual symptoms. Rarely, headache and vomiting may be signs of tumors involving the hypothalamus. Children usually do not complain of visual defects and NF1-OPGs can be unpredictable—as a result, a routine eye examination is required [26].
Although prognostic features are not clear, clinical experiences show that girls experience visual impairment more than boys. Additionally, postchiasmatic optic pathway-located OPGs and tumors that occur before the age of 2 appear to be more aggressive. After diagnosis, performing an eye examination and visual assessment with MRI is requested every 3 months in the first year for follow up.
Chemotherapy remains a mainstay treatment. Surgery seems to be problematic due to the location of these tumors and radiation is known to be a trigger for secondary malignancies along with vasculopathy. mTOR and MEK inhibitors for the treatment of optic pathway gliomas are still in progress [27].
References
- Gutmann, D.H.; Wood, D.L.; Collins, F.S. Identification of the neurofibromatosis type 1 gene product. Proc. Natl. Acad. Sci. USA 1991, 88, 9658–9662.
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369.
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, nrdp20174.
- Shah, K.N. The diagnostic and clinical significance of café-au-lait macules. Pediatr. Clin. N. Am. 2010, 57, 1131–1153.
- National Institutes of Health Consensus Development Conference Statement. Neurofibromatosis. Arch. Neurol. 1988, 45, 575–578.
- Nunley, K.S.; Gao, F.; Albers, A.C.; Bayliss, S.J.; Gutmann, D.H. Predictive value of café au lait macules at initial consultation in the diagnosis of neurofibromatosis Type 1. Arch. Dermatol. 2009, 145, 883–887.
- Pemov, A.; Sung, H.; Hyland, P.L.; Sloan, J.L.; Ruppert, S.L.; Baldwin, A.M.; Boland, J.F.; Bass, S.E.; Lee, H.J.; Jones, K.M.; et al. Genetic modifiers of neurofibromatosis Type 1-associated café-au-lait macule count identified using multi-platform analysis. PLoS Genet. 2014, 10, e1004575.
- Friedman, J.M.; Birch, P.H. Type 1 neurofibromatosis: A descriptive analysis of the disorder in 1728 patients. Am. J. Med. Genet. 1997, 70, 138–143.
- Riccardi, V.M. Pathophysiology of neurofibromatosis. J. Am. Acad. Dermatol. 1980, 3, 157–166.
- De Schepper, S.; Maertens, O.; Callens, T.; Naeyaert, J.-M.; Lambert, J.; Messiaen, L. Somatic mutation analysis in NF1 café au lait spots reveals two NF1 hits in the melanocytes. J. Investig. Dermatol. 2008, 128, 1050–1053.
- De Schepper, S.; Boucneau, J.; Haeghen, Y.V.; Messiaen, L.; Naeyaert, J.-M.; Lambert, J. Café-au-lait spots in neurofibromatosis type 1 and in healthy control individuals: Hyperpigmentation of a different kind? Arch. Dermatol. Res. 2006, 297, 439–449.
- Diwakar, G.; Zhang, D.; Jiang, S.; Hornyak, T.J. Neurofibromin as a regulator of melanocyte development and differentiation. J. Cell Sci. 2008, 121, 167–177.
- Evans, D.G.; E Baser, M.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314.
- Leroy, K.; Dumas, V.; Martin-Garcia, N.; Falzone, M.C.; Voisin, M.C.; Wechsler, J.; Revuz, J.; Créange, A.; Levy, E.; Lantieri, L.; et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: A clinicopathologic and molecular study of 17 patients. Arch. Dermatol. 2001, 137, 908–913.
- Coffin, C.M.; Cassity, J.; Viskochil, D.; Randall, R.L.; Albritton, K. Non-neurogenic sarcomas in four children and young adults with neurofibromatosis type 1. Am. J. Med. Genet. 2004, 40–43.
- Prada, C.E.; Rangwala, F.A.; Martin, L.J.; Lovell, A.M.; Saal, H.M.; Schorry, E.K.; Hopkin, R.J. Pediatric plexiform neurofibromas: Impact on morbidity and mortality in neurofibromatosis Type 1. J. Pediatr. 2012, 160, 461–467.
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Granada, C.N.P.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232.
- Farid, M.; Demicco, E.G.; A Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201.
- Hirbe, A.C.; Cosper, P.F.; Dahiya, S.; Van Tine, B.A. Neoadjuvant ifosfamide and epirubicin in the treatment of malignant peripheral nerve sheath tumors. Sarcoma 2017, 2017, 1–6.
- Sobczuk, P.; Teterycz, P.; Czarnecka, A.M.; Świtaj, T.; Kosela-Paterczyk, H.; Kozak, K.; Falkowski, S.; Rutkowski, P. Systemic treatment for advanced and metastatic malignant peripheral nerve sheath tumors—A sarcoma reference center experience. J. Clin. Med. 2020, 9, 3157.
- Manji, G.A.; Van Tine, B.A.; Lee, S.M.; Raufi, A.; Patwardhan, P.; Blumberg, L.E.; Sender, N.; Wang, J.; Otap, D.; Singh-Kandah, S.V.; et al. Phase 1 combination therapy with pexidartinib (PEX) and sirolimus (S) to target tumor-associated macrophages in pigmented villonodular synovitis, malignant peripheral nerve sheath tumors, and other soft tissue sarcomas. J. Clin. Oncol. 2019, 37 (Suppl. 15), 11055.
- Özdemir, B.C.; Bohanes, P.; Bisig, B.; Missiaglia, E.; Tsantoulis, P.; Coukos, G.; Montemurro, M.; Homicsko, K.; Michielin, O. Deep response to anti-PD-1 therapy of metastatic neurofibromatosis type 1-associated malignant peripheral nerve sheath tumor with CD274/PD-L1 amplification. JCO Precis. Oncol. 2019, 1–6.
- Prudner, B.C.; Ball, T.; Rathore, R.; Hirbe, A.C. Diagnosis and management of malignant peripheral nerve sheath tumors: Current practice and future perspectives. Neuro-Oncol. Adv. 2019, 2 (Suppl. 1), i40–i49.
- Guillamo, J.; Créange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic factors of CNS tumours in neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 2002, 126, 152–160.
- Lewis, R.A.; Gerson, L.P.; Axelson, K.A.; Riccardi, V.M.; Whitford, R.P. Von Recklinghausen neurofibromatosis. Ophthalmology 1984, 91, 929–935.
- Listernick, R.; Ferner, R.E.; Liu, G.T.; Gutmann, D.H. Optic pathway gliomas in neurofibromatosis-1: Controversies and recommendations. Ann. Neurol. 2007, 61, 189–198.
- Campen, C.J.; Gutmann, D.H. Optic pathway gliomas in neurofibromatosis type 1. J. Child Neurol. 2018, 33, 73–81.


