 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Selvakumar Subbian | + 2479 word(s) | 2479 | 2021-01-25 04:31:08 | | | |
| 2 | Camila Xu | Meta information modification | 2479 | 2021-02-02 05:30:22 | | | | |
| 3 | Selvakumar Subbian | Meta information modification | 2479 | 2021-06-03 15:22:31 | | | | |
| 4 | Camila Xu | Meta information modification | 2479 | 2021-06-04 03:06:30 | | |
Video Upload Options
The interferon (IFN) family of cytokines is broadly classified into type I, II, and III. The IFN-I consists of 13 subtypes, including IFNα, β, κ, ε, ω, and τ, all of which bind to the interferon-alpha receptor complex (IFNAR), composed of the IFNARI and IFNARII chains.
1. Type-I IFN Signaling
Plasmacytoid dendritic cells (DC) are the primary producers of IFNα, and binding of IFNα to IFNAR activates the JAK-STAT signaling pathway that induces interferon-stimulated genes (ISGs) [4]. Together, the JAK-STAT pathway and ISG molecules play crucial roles in the host immune response to bacterial, viral, and fungal diseases and associated inflammation [4]. Pathogen recognition by phagocytes through pathogen-associated molecular patterns (PAMPs) activates multiple and divergent host immune response signaling networks, including the IFN-I response pathways. Subsequent induction of downstream ISGs through the IFN-I pathway facilitates efficient viral killing and controls inflammation. During SARS-CoV-2 infection, the host cytoplasmic molecules, such as retinoic acid-inducible gene-1 (RIG-1) and melanoma differentiation-associated gene-5 (MDA5), recognize intracellular viral RNA and activate the IFN-I pathway, which ultimately enhances the production of effector molecules such as ISGs. Since ISGs regulate a vast range of cellular functions, ranging from transcription to translation of proinflammatory and metabolic molecules in phagocytic cells, any impairment in IFN-I signaling can be harmful to the host. Thus, optimal and timely activation of IFN-I signaling is crucial for a well-coordinated antiviral effect of phagocytes with minimal damage to the host [1][2][3][4].
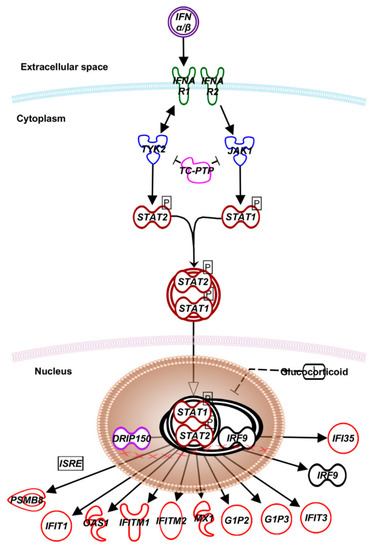
Figure 1. Canonical type I interferon signaling. IFN-I family of cytokines, encompassing IFN-α, -β, and –κ play critical roles in initiating, maintaining, and regulating host immune responses, particularly against viral infections [5][6]. The IFN signaling starts with type I and II IFN receptors (IFNR), which are transmembrane proteins that function as heterodimers. While type I IFNR comprises AR1 and AR2 subunits, type II IFNR is made of GR1 and GR2. The IFNAR1 and IFNGR1 are involved in binding type I and type II IFNs, respectively, while IFNAR2 and IFNGR2 subunits are involved in signal transduction. Engagement of IFNAR2 and IFNGR2 activates tyrosine kinases JAK1, JAK2, and TYK2 that phosphorylate the STAT1 and 2 proteins. After dimerization, the STAT proteins translocate to the nucleus. In the case to type I IFNs, the STAT1/2 heterodimers associate with IFN gene regulatory factor-9 (IRF-9), forming ISGF3, a trimeric transcription factor. The vitamin D receptor-interacting protein150 (DRIP150) interacts with STAT2 and stimulates IFN-dependent transcription. ISGF3 and DRIP150 complex bind to the IFN-stimulated regulatory elements (ISREs) to induce the expression of several IFN-α/β-regulated genes [5][6]. Abbreviations: TYK2—tyrosine kinase 2; JAK1—Janus kinase 1; TC-PTP—T-cell protein tyrosine phosphatase; STAT—signal transducer and activator of transcription; IFI—interferon-induced protein; DRIP150—vitamin D receptor-interacting protein 150; IRF—interferon regulatory factor; PSMB—proteasome 20S subunit beta; IFIT—interferon-induced protein with tetratricopeptide repeats; OAS—O-acetyl serine; MX1—MX dynamin-like GTPase 1; G1P2—ISG-15 ubiquitin-like modifier; ISRE—interferon-stimulated response element.
Apart from ISGs, IFN-I augments host protection against viral infections by recruiting and activating innate (DC, natural killer (NK) cells, neutrophils, monocytes, and macrophages) and adaptive (T and B) immune cells [5][6].
2. Type-I IFN Response and IFN-I Immunotherapy for COVID-19
The IFN-I pathway also plays a central role in the pathogenesis of COVID-19 [4]. Perturbations in IFN-I and ISG levels are among the primary indicators of disease severity in COVID-19 cases. A significant reduction in serum IFN-I levels has been reported in critically ill COVID-19 patients with a high viral load in their respiratory tract than those with mild or moderate disease symptoms. Consistently, the number of plasmacytoid DC, the primary producers of IFNα, was reduced in patients with severe COVID-19, contributing to reduced IFN-I levels. However, data from SARS-CoV-2 infection studies conducted on lung epithelial cells expressing angiotensin-converting enzyme-2 (ACE2) receptor, as well as in human organoids, human bronchial cells, and in post-mortem lung samples of COVID-19 cases showed a moderate-to-high level of ISGs (e.g., MX1, MX2, OAS1, IFI6, IFI27, IFIT1, IFIT3, IFITM1, IFITM2, and IFITM3) despite a low-to-undetectable IFN-I level. This observation suggests that either the infected host cells are refractory to the COVID-19 defense mechanism or the uninfected cells also contribute to elevated ISG level (bystander effect).
Induction of ISGs in infected cells mounts a robust antiviral response against SARS-CoV-2, including blocking viral replication. Besides, upregulation of ISGs, such as IFIT and IFITM family genes, including OASL, OAS2, IFI6, MX1, and MX2, as well as proinflammatory cytokines, including IL-1β IL-10, TNF-α, IL-6, and IL-1Ra was noted in the bronchoalveolar lavage fluid of COVID-19 patients [7][8][9]. The expression level of several proinflammatory genes involved in IFN-I signaling, including MX1, STAT1, STAT2, IFITM1, IFIT2, IFI35, IFIH1, IRF7, and IFI16 was enriched in COVID-19 patients [10]. Furthermore, these molecules’ expression patterns could differentiate mild/moderate COVID-19 cases from healthy individuals [10]. However, due to the complexity of the inflammatory cascade associated with COVID-19 pathogenesis and the heterogeneity in disease presentation, it is challenging to associate specific inflammatory molecules’ gene expression patterns as surrogates to determine the mortality index of a given patient. Given the limited number of patient samples used in gene expression studies, it is premature to conclusively define biomarkers of disease severity in COVID-19, based purely on the expression pattern of a specific set of genes. Thus, extensive profiling of enriched inflammatory gene expression from different clinical samples of many patients with various disease severity is required to define and correlate biomarkers with corresponding prognoses of COVID-19 cases. A recent study on the transcriptome of lung epithelial cells and the pulmonary granulomas of non-human primates reported that ACE2, the primary receptor for SARS-CoV-2 entry into host cells, is also a member of ISG. This study also indicated a dual role for IFN-specific upregulation of ACE2 in tissue tolerance and infection upon SARS-CoV-2 entry into host cells [11]. Although induction of ISGs contributes to host cells’ antiviral effects against SARS-CoV-2, the mechanistic link between IFN-I, ISG, and ACE2 pathways and associated regulation of host immune response in COVID-19 remains unknown.
Comorbidities and genetic susceptibility of the host can also alter the IFN-I response to viral infections and cause detrimental effects to the host [10]. During the initial stages of COVID-19, when the viral load, disease severity, and antibody responses are low, IFN-I plays a beneficial host effect by inhibiting viral propagation in the host. Consistently, elevated levels of IFN-I were observed at the infection site and in the blood of COVID-19 cases with mild symptoms and low viral burden. Thus, elevated IFN-I appears to be a host response against SARS-CoV-2 replication, dampening inflammation and alleviating disease symptoms. However, in COVID-19 patients with severe disease symptoms and a higher viral load, including the elderly population and those with comorbid conditions, the expression of IFN-I was significantly suppressed and was associated with exacerbated tissue inflammation and disease pathology [12]. Notably, the causal link between the level of IFN-I in the host and the severity of COVID-19 remains unknown. Similarly, the network/pathway regulating IFN-I-mediated antiviral and anti-inflammatory activities during COVID-19-mediated tissue damage is yet to be determined.
Several IFN-I subtypes, particularly IFN-α and IFN-β, are currently evaluated in several clinical trials for their therapeutic potential in controlling COVID-19 (Table 1). The rationale for IFN-I therapy is that compensating for the loss of IFN-I during SARS-CoV-2 would aid in viral clearance and alleviate inflammation associated with the disease severity among COVID-19 cases. However, in light of its dual and opposing role of IFN-I in COVID-19 pathogenesis, the use of IFN-I treatment should be carefully considered. Most of these clinical studies are either recruiting or yet to recruit patients for evaluating the beneficial effect of various IFN-I immunotherapy against COVID-19. Availability of data from these trials would inform about the efficacy of each of these IFN-I sub-types and factors impacting IFN-I immunotherapy for COVID-19 cases in the future.
Table 1. Description of various clinical trials testing IFN-I therapy for COVID-19 $.
| Trial ID | IFN-I Molecule | Adjunctive Drug # | Route of IFN-I Administration | Study Sponsor | Reference/Status * |
|---|---|---|---|---|---|
| ChiCTR2000029387 | IFNα1β | Lopinavir/Ritonavir or Ribavirin | Subcutaneous | Chongqing Public Health Medical Center, China | Recruiting |
| ChiCTR2000029638 | IFNα (“super-compound”) | Umifenovir or Lopinavir and Ritonavir | Nebulization | West China Hospital, China | Recruiting |
| ChiCTR2000030262 | IFNκ | Trefoil Factor Family 2 | Aerosol inhalation | Shanghai Medical College, China | Recruiting |
| ChiCTR2000030013 | IFNα1β | Standard care | Nasal spray | Chinese PLA General Hospital, China | Not recruiting |
| ChiCTR2000030166 | IFNα2β | Lopinavir/Ritonavir or Qing-Wen Bai-Du-Yin granules | Injection (route not disclosed) | The 5th Medical Center Chinese PLA General Hospital, China | Not recruiting |
| ChiCTR2000030922 | IFNα2α | Ribavirin | Route not disclosed | Foshan First People’s Hospital, China | Recruiting |
| ChiCTR2000029989 | IFNα1β | Standard care | Eye drops | Chinese PLA General Hospital, China | Not recruiting |
| ChiCTR2000030082 | IFNα | Dihydroartemisinin/Piperaquine or Ardibdol | Route not disclosed | The First Affiliated Hospital of Nanchang University, China | Suspended |
| ChiCTR2000029600 | IFNα | Lopinavir/Ritonavir or Favipiravir | Atomization (nasal spray) | The Third People’s Hospital of Shenzhen, China | Recruiting |
| ChiCTR2000030535 | IFNα | Ebastine and Lopinavir | Aerosol inhalation | Mianyang Central Hospital, China | Recruiting |
| IRCT20100228003449N27 | IFNβ1α | Hydroxychloroquine, Lopinavir/Ritonavir | Subcutaneous | Tehran University of Medical Sciences, Iran | Recruiting |
| IRCT20100228003449N28 | IFNβ1α | Hydroxychloroquine, lopinavir/ritonavir | Subcutaneous | Tehran University of Medical Sciences, Iran | Recruiting |
| 2020-001023-14 | IFNβ1α | Standard care | Aerosol inhalation | Synairgen Ltd., United Kingdom | Recruiting |
| 2020-001113-21 (RECOVERY Trial) | IFNβ1α | Standard care | Aerosol inhalation | University of Oxford, United Kingdom | Recruiting |
| 2020-000936-23 (DisCoVeRy Trial) | IFNβ1α | Standard care | Subcutaneous | INSERM, France | Recruiting |
| NCT04315948 (DisCoVeRy Trial) | IFNβ1α | Lopinavir/Ritonavir or Standard care | Subcutaneous | Institut National de la Sante Et de la Recherche Medicale, France | Recruiting |
| NCT04276688 | IFNβ1β | Lopinavir/Ritonavir, Ribavirin | Subcutaneous | The University of Hong Kong, Hong Kong. | Recruitment completed |
| NCT04254874 | IFNα2β | Abidol hydrochloride | Atomization (nasal spray) | Tongji Hospital, China | Recruiting |
| NCT04293887 | IFNα1β | Lopinavir/Ritonavir, Remdesivir | Nebulization inhalation | Tongji Hospital, China | Not recruiting |
| NCT04552379 | IFNβ1α | Standard care | Subcutaneous | Pontificia Universidad Catolica de Chile | Recruiting |
| NCT04273763 | IFNα2β | Bromhexine Hydrochloride, Abidol hydrochloride and Standard care | Atomization (nasal spray) | WanBangDe Pharmaceuticals, China | Not recruiting |
| NCT04291729 | IFNα2 | Danoprevir/Ritonavir | Spray inhalation | The Ninth Hospital of Nanchang, China | Completed |
| NCT04251871 | IFNα | Lopinavir/Ritonavir or Traditional Chinese Medicines | Aerosol inhalation | Beijing 302 Hospital, China | Recruiting |
| NCT04275388 | IFNα | Lopinavir/Ritonavir and Abidor Hydrochloride | Nebulization inhalation | Jiangxi Qingfeng Pharmaceutical Co. Ltd. | Not recruiting |
| NCT04469491 | IFNβ1β | Standard care | Nebulization inhalation | Centre Hospitalier Universitaire, Amiens | Suspended |
$ Data obtained from the WHO COVID-19 Vaccine website [1]. * status date was current as of 3 January 2021. # some studies involve “Standard care”, which includes treatment with/without antibiotics, corticosteroids or hydroxychloroquine and oxygen support, in addition to IFN-I therapy. ChiCTR—Chinese Clinical Trial Registry; IRCT—Iranian Registry of Clinical Trials; RECOVERY—Randomized Evaluation of COVID-19 Therapy; DisCoVeRy—Trial of Treatments for COVID-19 in Hospitalized Adults; NCT—National Clinical Trial.
3. Implications of IFN-I Immunotherapy on the Host Response to Other Infectious Diseases
Below is a summary of health conditions in which IFN-I immunotherapy can potentially impact the host response to bacterial and viral infections and the clinical outcome of treatment.
3.1. Viral Diseases
IFN therapy has been credited as prophylaxis to prevent virus-induced exacerbations in airway diseases, such as chronic obstructive pulmonary disease (COPD). A recent report using the primary cell culture model of respiratory syncytial virus (RSV) and influenza virus infection shows that intermittent treatment with IFN-β was beneficial to the host cells and did not trigger proinflammatory cytokines, such as IL-1β, TNF, and IL-6 [13]. This observation suggests that IFN-I therapy helps to prevent COVID-19-associated pulmonary exacerbations in patients with pre-existing COPD or co-infected with other viral pathogens such as RSV.
The role of IFN-I in human immunodeficiency virus (HIV) infection and its therapeutic role is inconclusive. Upon the entry of HIV-1, the pathogen recognition receptors (PRR) on phagocytes, including Toll-like receptor-7 (TLR7), cGAS, and IFI16, recognize the viral antigens and secrete IFN-I. The binding of IFN-I to IFNAR and phosphorylation of the downstream JAK/STAT pathway leads to the upregulation of ISGs, which are proven antiviral defense molecules (Figure 1). Since the induction of ISGs can deplete CD4+ and CD8+ cells, a low level of IFN-I during the early stages of HIV-1 infection was presumed beneficial. Thus, a delayed and/or sub-optimal IFN-I production can lead to an immunosuppressive niche and persistent, chronic disease. However, during latent HIV-1 infection, sustained IFN-I signaling is associated with immune activation and viral persistence. In summary, IFN-I signaling has a paradoxical yet dynamic role in the pathogenesis of acute vs. chronic/persistent HIV-1 infection [14][15]. Hence, caution should be taken when administering IFN-I treatment to COVID-19 patients who are also HIV-positive. Clinical parameters, such as blood CD4+ T cell counts and co-infection with other pathogens, should be monitored in these patients during IFN therapy.
3.2. Nosocomial Infection
Reports have shown that COVID-19 cases acquire secondary nosocomial infections, including multidrug-resistant Staphylococcus aureus and Streptococcus pneumonia, possibly due to lower IFN-I levels in these patients. However, excessive IFN-I levels have been associated with increased susceptibility to disease progression with worsened pneumonia. Moreover, elevated IFN-I molecules inhibit the proinflammatory TH17 signaling and impair the recruitment of macrophages and neutrophils, as well as neutrophil chemokine secretion, all of which facilitate the progression of nosocomial infection [16]. However, the correlation between IFN-I levels and disease severity in COVID-19 or secondary nosocomial infections is not fully understood. Since critically ill COVID-19 patients in hospitals are more prone to nosocomial infections, the dose and route of administration of IFN-I and therapy duration should be planned cautiously.
3.3. Tuberculosis
Upregulation of IFN-I pathway molecules has been reported during infection by Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), a contagious bacterial disease marked by severe, chronic lung inflammation [17]. TB, primarily a pulmonary disease, caused 1.5 million deaths worldwide and an estimated 10 million new cases in 2018 (https://www.who.int/tb/publications/factsheet_global.pdf?ua=1). Elevated IFN-I levels correlate with the disease severity caused by hypervirulent Mtb strains and increased host susceptibility to active TB. Several possible mechanisms, including suppression of proinflammatory innate and Th1 responses, early cell death of alveolar macrophages, recruitment of permissive myeloid cells, and the reduced macrophage antibacterial responses, are associated with the detrimental effect of elevated IFN-I in Mtb-infected host [17]. However, when co-administered with anti-mycobacterial therapy or Bacille Calmette-Guerin (BCG) vaccine, IFN-α conferred host-protective effect against Mtb infection. Studies using human PBMCs and animal models showed that priming with IFN-α increases the BCG vaccine’s efficacy and promotes T-cell priming and IFN-γ-induced production of “host-protective” proinflammatory cytokines, including IL-12, TNF-α, IL-17, and IL1β [18]. Notably, the role of IFN-I in regulating chronic, asymptomatic latent Mtb infection (LTBI) and their potential for reactivating LTBI to active TB disease remains unclear. Thus, IFN-I treatment of COVID-19 cases with underlying LTBI might lead to TB reactivation. Moreover, patients with symptomatic, active TB have elevated IFN-I levels [17]. Therefore, in the context of Mtb infection, the effect of IFN-I response can be beneficial or detrimental to the host, dependent on whether the host is immunized with BCG or having LTBI/active TB, respectively. In this regard, measuring the IFN-I level in samples, and TB or LTBI status of the patient, determined by skin test or quantiferon test or chest X-ray, would be valuable for initiating IFN therapy for COVID-19 cases. This approach would alleviate the risk for reactivation of LTBI or exacerbation of TB among COVID-19 patients.
References
- WHO. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/. (accessed 7 November 2020 and 8 January 2021).
- Khalaf, K.; Papp, N.; Chou, J.T.; Hana, D.; Mackiewicz, A.; Kaczmarek, M. SARS-CoV-2: Pathogenesis, and Advancements in Diagnostics and Treatment. Front. Immunol. 2020, 11, 570927.
- U.S. National Library of Medicine. Clinical Trials–COVID19. Available online: https://www.clinicaltrials.gov/ct2/results?cond=COVID-19. (accessed on 7 November 2020).
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103.
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061.
- Teijaro, J.R. Pleiotropic Roles of Type 1 Interferons in Antiviral Immune Responses. Adv. Immunol. 2016, 132, 135–158.
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog 2020, 16, e1008737.
- Zhou, R.; To, K.K.; Wong, Y.C.; Liu, L.; Zhou, B.; Li, X.; Huang, H.; Mo, Y.; Luk, T.Y.; Lau, T.T.; et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity 2020, 53, 864–877 e5.
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890 e2.
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et. al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724.
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J., Hauser, B.M.; et. al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035 e19.
- Park, A.; Iwasaki, A. Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878.
- Johnston, S.L. IFN Therapy in Airway Disease: Is Prophylaxis a New Approach in Exacerbation Prevention? Am. J. Respir. Crit. Care Med. 2020, 201, 9–11.
- Scagnolari, C.; Antonelli, G. Type I interferon and HIV: Subtle balance between antiviral activity, immunopathogenesis and the microbiome. Cytokine Growth Factor Rev. 2018, 40, 19–31.
- Sugawara, S.; Thomas, D.L.; Balagopal, A. HIV-1 Infection and Type 1 Interferon: Navigating Through Uncertain Waters. AIDS Res. Hum. Retroviruses 2019, 35, 25–32.
- Parker, D. Impact of Type I and III Interferons on Respiratory Superinfections Due to Multidrug-Resistant Pathogens. J. Infect. Dis. 2017, 215, S58–S63.
- Moreira-Teixeira, L.; Mayer-Barber, K.; Sher, A.; O’Garra, A. Type I interferons in tuberculosis: Foe and occasionally friend. J. Exp. Med. 2018, 215, 1273–1285.
- Rivas-Santiago, C.E.; Guerrero, G.G. IFN-alpha Boosting of Mycobacterium bovis Bacillus Calmette Guerin-Vaccine Pro-moted Th1 Type Cellular Response and Protection against M. tuberculosis Infection. Biomed. Res. Int. 2017, 2017, 8796760.


