 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexander Ou | + 2451 word(s) | 2451 | 2021-01-18 07:30:55 | | | |
| 2 | Catherine Yang | Meta information modification | 2451 | 2021-01-27 04:14:22 | | |
Video Upload Options
Glioblastoma is the most common malignant primary brain tumor in adults and is almost invariably fatal. Despite our growing understanding of the various mechanisms underlying treatment failure, the standard-of-care therapy has not changed over the last two decades, signifying a great unmet need.
1. Introduction
Glioblastoma (GBM) is the most common malignant primary brain tumor in adults, and despite standard-of-care multimodality therapy, including maximal safe resection, radiotherapy, and chemotherapy, the prognosis remains almost universally fatal with a mean overall survival of 14 to 20 months [1]. Since the 2005 pivotal phase III trial by Stupp et al. [1], which established the role of concurrent chemoradiation with temozolomide followed by adjuvant temozolomide for patients with newly diagnosed glioblastoma, no chemotherapies investigated in late-phase clinical trials have significantly improved upon this foundational approach. The U.S. Food and Drug Administration (FDA) has approved the anti-vascular endothelial growth factor (VEGF) antibody bevacizumab for treatment of recurrent glioblastoma on the basis of two phase II studies showing a progression-free survival benefit. However, two phase III clinical trials evaluating its role in the treatment of newly diagnosed disease did not demonstrate an overall survival benefit when bevacizumab was added to standard therapy [2][3][4]. The FDA also has approved tumor-treating fields therapy (TTF), which consists of low-intensity, alternating electric fields applied to the scalp for most of the day, for use in recurrent (2011) and newly diagnosed (2015) glioblastoma, although widespread adoption of TTF has been limited by methodological concerns about the generalizability of the data from prior studies of it [5]. Thus, there are currently no effective therapies for glioblastoma. In this review, we discuss the innate mechanisms of treatment resistance common to all glioblastomas before characterizing the various mechanisms of resistance to conventional treatments, targeted therapies, and immunotherapy.
2. General Mechanisms of Treatment Resistance
2.1. The Blood–Brain and Blood–Brain–Tumor Barriers
The initial obstacle that therapies against malignant gliomas must overcome is the blood-brain barrier (BBB), a non-fenestrated physical barrier comprised of specialized capillary endothelial cells interconnected by multi-protein tight junctions consisting of claudins (especially claudin-1, -3, and -5), occludins, and junctional adhesion molecules [6]. Closely associated with these endothelial cells by virtue of a shared basal lamina are complexes of astrocytic endfeet, pericytes, and intermittent ends of neurons, which collectively constitute the neurovascular unit responsible for maintaining biochemical and physical homeostasis in the normal brain [7]. The BBB permits only small (<500 Da and <400 nm) and lipophilic molecules to passively diffuse across; other molecules cross the BBB via pinocytosis, receptor- or carrier-mediated transcytosis, and solute-carrier-protein mechanisms [8]. The integrity of the BBB and homeostatic equilibrium are further bolstered by ATP-binding cassette transporters, such as multidrug resistance-1 (MDR1), P-glycoprotein, breast cancer resistance protein, and numerous other drug resistance proteins that are expressed on the luminal and abluminal sides of vessel walls (Figure 1). These transporters actively mediate the efflux of xenobiotics such as cytotoxic or targeted therapeutic agents out of the brain parenchyma [9][10][11]. Attempts to modulate these efflux pumps have largely been unsuccessful [12][13].
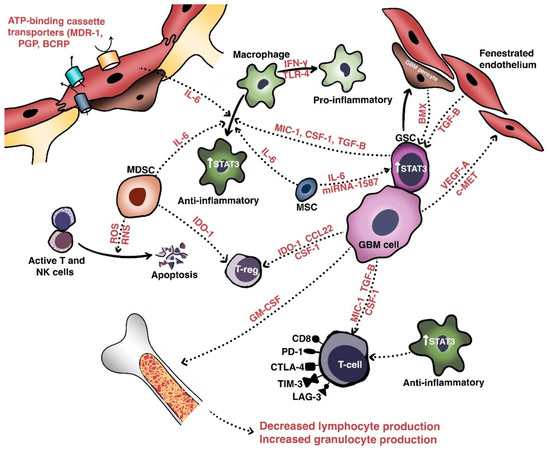
In glioblastoma and other high-grade intracranial neoplasms, the BBB is heterogeneously disrupted to form the blood–tumor–brain barrier, which is characterized by abnormal pericyte distribution, reduced tight junctions, the loss of astrocytic endfeet, and increased permeability to circulating immune cells [14][15]. This heterogeneity of tumoral vasculature creates regional niches of variable permeability to blood, oxygen, nutrients, and drugs. Recent work has further extended our understanding of a glioblastoma tumor’s centrally necrotic, hypoxic core, and less-hypoxic periphery [16]. Indeed, hypoxic glioblastoma cells secrete VEGF-A via exosomes to promote the proliferation of endothelial cells with downregulated expression of key junctional proteins such as claudin-5 and occludin [17][18].
Glioma stem cells (GSCs), which are pluripotent, slowly dividing, and therapy-resistant cells residing in the perivascular hypoxic niches of the brain, have been recognized for their importance in resisting cytotoxic therapies [19][20]. GSCs are not only intrinsically resistant to therapy but also exert substantial effects on neighboring cells within the microenvironment to maintain their populations [21]. In particular, glioblastoma pericytes derive from GSCs via trans-differentiation and contribute to the integrity of the BBB via the overexpression of proteins such as bone marrow and X-linked (BMX) non-receptor tyrosine kinase (Figure 1), which activate signaling through signal transducer and activator or transcription 3 (STAT3) to maintain the self-renewal capability of the GSCs occupying perivascular niches [14][22]. Indeed, in an orthotopic xenograft glioblastoma model, Zhou et al. [23] found that pericyte coverage not only correlated with the prognosis of patients with glioblastoma but also that inhibition of BMX with ibrutinib selectively disrupted the permeability of the blood–brain–tumor barrier and enhanced delivery of chemotherapy (e.g., etoposide) that ordinarily penetrates the BBB poorly, thus prolonging mouse survival [24].
The two most important signaling pathways involved in the formation of the BBB and the regulation of its integrity are the Wingless-related integration site (Wnt) and Sonic hedgehog (Shh) pathways. During normal embryonic development and in adulthood, Shh secreted by astrocytes binds to the patched-1 (Ptch-1) protein on endothelial cells or pericytes to activate the smoothened (Smo) protein. This leads to downstream transcriptional activation of genes bound by the Gli family of transcription factors, such as SOX-18 and TAL-1, which increase transendothelial resistance and decrease permeability by enhancing claudin expression [25][26][27]. Shh signaling also contributes to the central nervous system’s immune privilege by decreasing the expression of the intercellular adhesion molecule ICAM-1 and the secretion of chemokines such as CXCL8/interleukin (IL)-9 and CCL2/MCP-1 by endothelial cells. Similarly, Wnt/β-catenin signaling in endothelial cells contributes to the regulation of the BBB. The endothelial G-protein coupled receptor Gpr124 is one such crucial coactivator of Wnt7a and Wnt7b-stimulated canonical signaling via the binding of Frizzled receptor and Lrp coreceptor. Gpr124 upregulates claudin-5 expression, decreases platelet-derived growth factor receptor (PDGFR)-B expression, and increases pericyte coverage [28]. Recently, Griveau et al. [29] extended the understanding of glioma cellular phenotypes vis-à-vis the tumoral microenvironment, demonstrating in a mouse model that Olig2+/Wnt7+ glioma cells—analogous to the oligodendrocytes comprising the leading edge of glioblastoma tumors—invaded the brain parenchyma via co-option of blood vessels by single-cells, while Olig2-/Wnt7- glioma cells—analogous to proneural glioblastoma cells—proliferated in the perivascular niche and expressed abundant VEGF-C and VEGFR-1/2/3 to form dense tumor collections with leaky vasculature. Importantly, anti-angiogenic therapy (i.e., VEGF inhibition) led to selective enrichment of the Olig2+/Wnt7+ cells, indicating a mechanism through which glioblastoma cells may ultimately overcome prolonged anti-angiogenic therapy.
Strategies to breach the BBB and improve drug delivery have therefore focused on mechanical disruption (i.e., osmotic disruption) and invasive local delivery (e.g., convection-enhanced delivery), and these strategies have been limited by either unacceptable toxicity or inefficacy [30][31][32]. Focused ultrasound is a relatively new modality which transiently renders the BBB permeable to allow for improved drug delivery with a more favorable adverse effect profile, and clinical study is underway [33]. Continued efforts to improve drug delivery via nanoparticle- or peptide-based drug-carrying methods are ongoing, and further study of pharmacological inhibition of Wnt/Shh signaling and pericyte function is warranted [34][35].
2.2. Intra- and Intertumoral Heterogeneity
Perhaps the most important and challenging barrier to establishing effective treatments for glioblastoma is tumoral heterogeneity, which encompasses a vast spectrum of molecular, genetic, cellular, temporal, spatial, and evolutionary diversity and prevents the use of any single universal therapeutic approach. The Cancer Genome Atlas (TCGA) Research Network originally used an extensive characterization of the genomic alterations in glioblastoma to identify three critical signaling pathways in the disease—p53, retinoblastoma (Rb), and receptor tyrosine kinase/Ras/PI3K—and subsequent groups have built upon these data to formulate classification schemes with prognostic importance. Verhaak et al. [36][37] used factor analysis and consensus clustering of data from TCGA to define 4 glioblastoma subtypes on transcriptional grounds: classical, mesenchymal, proneural, and neural. The classical subtype is characterized by the gain of chromosome 7 and loss of chromosome 10, epidermal growth factor receptor (EGFR) amplification, and cyclin dependent kinase inhibitor 2A (CDKN2A) homozygous deletion with high-level upregulation of Notch (NOTCH1, NOTCH3, JAG1, LFNG)- and Shh (SMO, GAS1, GLI2)-related signaling with downregulation of mitogen-activated protein kinase (MAPK) and pro-apoptotic proteins such as cleaved caspase 7 and 9, Bid, and Bak. The mesenchymal subtype is characterized by mutations in NF1, phosphatase, and tensin-homolog protein (PTEN), and the nuclear factor κ-light chain-enhancer of activated B cells (NF-κB) signaling pathway (e.g., TRADD, RELB, and TNFRSF1A) with increased MAPK and decreased mTOR signaling. The proneural subtype is characterized by IDH1 mutations, PDGFRA amplification, TP53 mutation, PI3K signaling, and high expression levels of oligodendrocytic developmental genes (e.g., OLIG2, NKDX2-2, and unique genomic hypermethylation-designated glioma-CpG island methylator phenotype or G-CIMP+) [38][39][40]. The genuine existence of the neural subtype is controversial, as subsequent studies by Wang et al. [41] and Gill et al. [42] have suggested that sampling of non-neoplastic cells at the infiltrative margins of the tumor account for the transcriptional profile was observed. Whole exome and transcriptional sequencing and proteomic profiling have further refined our understanding of the molecular subtypes. Various EGFR alterations (e.g., gene deletions or fusions) have also been detected, signifying the sheer genetic complexity of glioblastoma.
A model of cellular states and genetic diversity in glioblastoma recently postulated by Neftel et al. [43] integrated single-cell RNA sequencing and bulk genomic/transcriptomic and single-cell lineage tracing to demonstrate that sets of genes—designated “meta-modules”and encompassing mesenchymal, astrocytic, oligodendroglial, stem cell, and neural progenitor cell programs—recurred at high rates between tumors despite substantial intratumoral heterogeneity. Cellular populations isolated on the basis of these meta-modules generally expressed only 1 meta-module, and 15% of cells expressed 2, suggesting a hybrid subtype. Importantly, multiple cellular states were found to coexist within each tumor, each partially dictated by genetic mutations such as EGFR or PDGFRA.
It remains to be seen how this knowledge of the complex genomic, transcriptomic, epigenomic, and proteomic programs may be best applied to develop a therapeutic strategy to treat glioblastoma. However, it is clear from the information above that any approach must be individualized to some degree.
2.3. Heterogeneity of the Tumoral Microenvironment
Intratumoral heterogeneity is generated not only by the various populations of resident cells and their intercellular communications, but also by the unique niches created by the vasculature and extracellular matrix [20]. These aggregate populations and the cross-talk molecules they share are collectively termed the tumor microenvironment (TME). Relevant cellular populations of the TME (Figure 1) include glioblastoma/glioma stem cells, tumor-infiltrating lymphocytes (TILs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and BBB cells (e.g., endothelial cells and pericytes). Intercommunication between these populations of cells occurs via secreted factors with shared signaling pathways that mediate growth, invasion, immune escape, and therapeutic resistance.
As mentioned above, recent studies support the hypothesis that the TME is partially genetically driven. Indeed, proneural (i.e., PDGFB-mutated) tumors demonstrate more permeable BBBs than mesenchymal (i.e., NF1-mutated) tumors and IDHwt gliomas harbor more monocyte-derived macrophages than microglia relative to IDHmut gliomas [44][45][46]. Transcriptomic data further suggest that proneural cells are concentrated in the leading, infiltrative edges of tumors where they use creatinine to resist the formation of reactive oxygen species via a hypoxia-inducible factor-dependent mechanism [47]. Furthermore, regression analysis-based gene deconvolution of RNA sequencing data from TCGA has also demonstrated a significant association between mesenchymal or classical composition and higher macrophage content with more negative regulation of T-cell activation [48]. Mesenchymal cells also express high levels of caspase-8, which activates NF- κB signaling in a non-canonical pathway to increase angiogenesis, growth, and transcription of factors such as VEGF and IL-8 [49][50].
In the perivascular space, a major site of cross-talk within the TME, glioblastoma cells interact with components of the BBB to promote their own survival, growth, and immune escape. Hepatocyte growth factor (HGF) secreted by tumor cells binds with c-Met to induce the transformation of endothelial cells—via Wnt/β-catenin signaling—into mesenchymal populations. These mesenchymal populations form aberrant neovasculature to promote the invasion, proliferation, and generation of GSC-maintaining hypoxic milieus [51][52]. Pericytes also maintain the TME in the perivascular spaces, in which interaction with glioblastoma cells induces an oxidative burst that promotes upregulation of the lysosome-associated membrane protein 2A (LAMP-2A) and chaperone-mediated autophagy. This leads to secretion of anti-inflammatory cytokines (e.g., IL-10, TGF-β), increased programmed death-ligand 1 (PD-L1) expression, and decreased major histocompatibility complex-II and co-stimulatory molecule expression. The net effect is the promotion of immune tolerance [50][53].
In terms of immune-cell composition, glioblastoma includes both microglia and peripherally-recruited macrophages and a smaller number of TILs. Glioblastoma cells secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) to promote a shift in bone marrow hematopoiesis toward granulocytic lineages, and this causes a reduction in lymphocytic cells. Glioblastoma cells also secrete chemokines such as C-C motif chemokine 22 (CCL22) to promote regulatory T cell (Treg) infiltration [54][55][56]. TILs induce indoleamine 2,3 dioxygenase 1 (IDO1) expression in glioblastoma cells to promote CD25+/FoxP3+ Treg infiltration [50]. Macrophages are a major immunosuppressive cell population within the TME and have two phenotypes. The pro-inflammatory, immune-reactive phenotype is typically acquired after stimulation with toll-like receptor 4 ligands and interferon-γ, while the alternative anti-inflammatory, immune tolerant phenotype occurs after IL-4, IL-10, and IL-13 exposure and mediates immunosuppressive effects [57]. Recent work suggests that endothelial cells expressing IL-6 and microenvironmental colony-stimulating factor-1 (CSF-1) synergistically activate Akt/mTOR and contribute to a more immunosuppressive polarization [58].
Extracellular components also play an important role in maintaining the TME. To this end, glioblastoma cells secrete multiple types of molecules to enhance invasion via cell-matrix interactions, neovascularization, and growth [21][59]. One of these is tenascin-C, a glycoprotein that enhances the invasiveness of glioblastoma cells via non-adhesion and the focal-adhesion kinase pathway [60]. Fibulin-3 is another soluble glycoprotein secreted by glioblastomas that exerts effects on endothelial cells, astrocytes, and GSCs that promote growth, invasion, chemoresistance, and survival via both Notch- and NF-κB-dependent mechanisms [61][62][63]. In addition, exosomes (extracellular vesicles extruded by glioblastoma cells) contain fusion proteins that promote the mesenchymal transformation, stemness, and invasiveness of glioblastoma cells and endothelial neovascularization [64]. Recent work has identified another stromal population of human mesenchymal stem cells that secretes IL-6 and exosomal miR-1587 to promote GSC proliferation and stemness [65].
As discussed above, therapy-resistant cellular populations within the TME that promote angiogenesis, immunosuppression, and the maintenance of stemness contribute substantially to treatment resistance, and continued efforts to target these populations are warranted. The inhibition of one such axis, that of macrophage- and MDSC-related CSF-1 and its receptor, has shown promise in preclinical models of glioma, and further study is warranted to determine its therapeutic role [66][67].
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003, 5, 79–88.
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II Trial of Single-Agent Bevacizumab Followed by Bevacizumab Plus Irinotecan at Tumor Progression in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 740–745.
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. New Engl. J. Med. 2014, 370, 709–722.
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastomaa: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543.
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview: Structure, regulation, and clinical implica-tions. Neurobiol. Dis. 2004, 16, 1–13.
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412.
- Pappenheimer, J.R.; Renkin, E.M.; Borrero, L.M. Filtration, Diffusion and Molecular Sieving Through Peripheral Capillary Membranes; a con-tribution to the pore theory of capillary permeability. Am. J. Physiol. Content 1951, 167, 13–46.
- Ueda, K.; Cornwell, M.M.; Gottesman, M.M.; Pastan, I.; Roninson, I.B.; Ling, V.; Riordan, J.R. The mdrl gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem. Biophys. Res. Commun. 1986, 141, 956–962.
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; E Mackie, J.; E Grant, C.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line [published correction appears in Science]. Science 1992, 258, 1650–1654.
- Tsuji, A.; Terasaki, T.; Takabatake, Y.; Tenda, Y.; Tamai, I.; Yamashima, T.; Moritani, S.; Tsuruo, T.; Yamashita, J. P-glycoprotein as the drug efflux pump in primary cultured bovine brain capillary endothelial cells. Life Sci. 1992, 51, 1427–1437.
- Becker, C.M.; Oberoi, R.K.; McFarren, S.J.; Muldoon, D.M.; Pafundi, D.H.; Pokorny, J.L.; Brinkmann, D.H.; Ohlfest, J.R.; Sarkaria, J.N.; Largaespada, D.A.; et al. Decreased affinity for efflux transporters increases brain penetrance and molecular targeting of a PI3K/mTOR inhibitor in a mouse model of glioblastoma. Neuro Oncol. 2015, 17, 1210–1219.
- Kizilbash, S.H.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A.; et al. Restricted Delivery of Talazoparib Across the Blood–Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer 2017, 16, 2735–2746.
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152.
- Ratnam, N.M.; Gilbert, M.R.; Giles, A.J. Immunotherapy in CNS cancers: The role of immune cell trafficking. Neuro Oncol. 2018, 21, 37–46.
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410.
- Zhao, C.; Wang, H.; Xiong, C.; Liu, Y. Hypoxic glioblastoma release exosomal VEGF-A induce the permeability of blood-brain barrier. Biochem. Biophys. Res. Commun. 2018, 502, 324–331.
- Wang, W.; Dentler, W.L.; Borchardt, R.T. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am. J. Physiol. Circ. Physiol. 2001, 280, H434–H440.
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nat. Cell Biol. 2004, 432, 396–401.
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82.
- Mettang, M.; Meyer-Pannwitt, V.; Karpel-Massler, G.; Zhou, S.; Carragher, N.O.; Föhr, K.J.; Baumann, B.; Nonnenmacher, L.; Enzenmüller, S.; Dahlhaus, M.; et al. Blocking distinct interactions between Glioblastoma cells and their tissue microenvironment: A novel multi-targeted therapeutic approach. Sci. Rep. 2018, 8, 1–14.
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor Tyrosine Kinase BMX Maintains Self-Renewal and Tumorigenic Potential of Glioblastoma Stem Cells by Activating STAT3. Cancer Cell 2011, 19, 498–511.
- Zhou, W.; Chen, C.; Shideng, B.; Bian, X.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.-F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 2017, 21, 591–603.e4.
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10, eaah6816.
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731.
- Findley, M.K.; Koval, M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life 2009, 61, 431–437.
- Roudnicky, F.; Kim, B.K.; Lan, Y.; Schmucki, R.; Küppers, V.; Christensen, K.; Graf, M.; Patsch, C.; Burcin, M.; Meyer, C.A.; et al. Identification of a combination of transcription factors that synergistically increases endothelial cell barrier resistance. Sci. Rep. 2020, 10, 1–9.
- Chang, J.; Mancuso, M.R.; Maier, C.; Liang, X.; Yuki, K.; Yang, L.; Kwong, J.W.; Wang, J.; Rao, V.; Vallon, M.; et al. Gpr124 is essential for blood–brain barrier integrity in central nervous system disease. Nat. Med. 2017, 23, 450–460.
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.-C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889.e7.
- Desjardins, A.; Gromeier, M.; Ii, J.E.H.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. New Engl. J. Med. 2018, 379, 150–161.
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF- 2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro Oncol. 2010, 13, 132–142.
- Kunwar, S.; Chang, S.M.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010, 12, 871–881.
- Chen, K.-T.; Lin, Y.-J.; Chai, W.-Y.; Lin, C.-J.; Chen, P.-Y.; Huang, C.-Y.; Kuo, J.S.; Liu, H.-L.; Wei, K.-C. Neuronavigation-guided focused ultrasound (NaviFUS) for transcranial blood-brain barrier opening in recurrent glioblastoma patients: Clinical trial protocol. Ann. Transl. Med. 2020, 8, 673.
- Thomas, E.; Colombeau, L.; Gries, M.; Peterlini, T.; Mathieu, C.; Thomas, N.; Boura, C.; Frochot, C.; Vanderesse, R.; Lux, F.; et al. Ultrasmall AGuIX theranostic nanoparticles for vascular-targeted interstitial photodynamic therapy of glioblastoma. Int. J. Nanomed. 2017, 12, 7075–7088.
- Pandey, V.; Haider, T.; Chandak, A.R.; Chakraborty, A.; Banerjee, S.; Soni, V. Surface modified silk fibroin nanoparticles for improved delivery of doxorubicin: Development, characterization, in-vitro studies [published online ahead of print, 2020 August 3]. Int. J. Biol. Macromol. 2020, 164, 2018–2027.
- The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways [published correction appears in Nature]. Nat. Cell Biol. 2008, 455, 1061–1068.
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522.
- Kim, Y.-W.; Koul, D.; Kim, S.H.; Lucio-Eterovic, A.K.; Freire, P.R.; Yao, J.; Wang, J.; Almeida, J.S.; Aldape, K.; Yung, W.A. Identification of prognostic gene signatures of glioblastoma: A study based on TCGA data analysis. Neuro Oncol. 2013, 15, 829–839.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477.
- Wang, Q.; Hu, X.; Hu, B.; Muller, F.; Kim, H.; Squatrito, M.; Millelsen, T.; Scarpace, L.; Barthel, F.; Lin, Y.-H.; et al. Tumor evolution of glioma intrinsic gene expression subtype associates with immunological changes in the microenvironment. bioRxiv 2016, 052076.
- Gill, B.J.; Pisapia, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 12550–12555.
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21.
- Herting, C.J.; Chen, Z.; Pitter, K.L.; Szulzewsky, F.; Kaffes, I.; Kaluzova, M.; Park, J.C.; Cimino, P.J.; Brennan, C.; Wang, B.; et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017, 65, 1914–1926.
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17.
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20.
- Heiland, D.H.; Gaebelein, A.; Boerries, M.; Woerner, J.; Pompe, N.; Franco, P.; Heynckes, S.; Bartholomä, M.D.; Hailín, D.Ó.; Carro, M.S.; et al. Microenvironment-Derived Regulation of HIF Signaling Drives Transcriptional Heterogeneity in Glioblastoma Multiforme. Mol. Cancer Res. 2018, 16, 655–668.
- Luoto, S.; Hermelo, I.; Vuorinen, E.M.; Hannus, P.; Kesseli, J.; Nykter, M.; Granberg, K.J. Computational Characterization of Suppressive Immune Microenvironments in Glioblastoma. Cancer Res. 2018, 78, 5574–5585.
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Del Bufalo, D.; Cinà, I.; Di Benedetto, A.; Mottolese, M.; et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. eLife 2017, 6.
- Valdor, R.; García-Bernal, D.; Bueno, C.; Ródenas, M.; Moraleda, J.M.; Macian, F.; Martínez, S. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget 2017, 8, 68614–68626.
- Huang, M.; Liu, T.; Ma, P.; Mitteer, R.A.; Zhang, Z.; Kim, H.J.; Yeo, E.; Zhang, D.; Cai, P.; Li, C.; et al. c-Met–mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J. Clin. Investig. 2016, 126, 1801–1814.
- Huang, M.; Zhang, D.; Wu, J.Y.; Xing, K.; Yeo, E.; Li, C.; Zhang, L.; Holland, E.C.; Yao, L.; Qin, L.; et al. Wnt-mediated endothelial transformation into mesenchymal stem cell–like cells induces chemoresistance in glioblastoma. Sci. Transl. Med. 2020, 12, eaay7522.
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martínez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martínez, S. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665.
- Kast, R.E.; A Hill, Q.; Wion, D.; Mellstedt, H.; Focosi, D.; Karpel-Massler, G.; Heiland, T.; Halatsch, M.-E. Glioblastoma-synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: Potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumor Biol. 2017, 39.
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Győrffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660.
- Crane, C.A.; Ahn, B.J.; Han, S.J.; Parsa, A.T. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: Implications for immunotherapy. Neuro Oncol. 2012, 14, 584–595.
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555.
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 2018, 9, 559.
- Westhoff, M.-A.; Zhou, S.; Nonnenmacher, L.; Karpel-Massler, G.; Jennewein, C.; Schneider, M.; Halatsch, M.-E.; Carragher, N.O.; Baumann, B.; Krause, A.; et al. Inhibition of NF- B Signaling Ablates the Invasive Phenotype of Glioblastoma. Mol. Cancer Res. 2013, 11, 1611–1623.
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016, 18, 507–517.
- Nandhu, M.S.; Kwiatkowska, A.; Bhaskaran, V.; Hayes, J.; Hu, B.; Viapiano, M.S. Tumor-derived fibulin-3 activates pro-invasive NF-κB signaling in glioblastoma cells and their microenvironment. Oncogene 2017, 36, 4875–4886.
- Hu, B.; Nandhu, M.S.; Sim, H.; Agudelo-Garcia, P.A.; Saldivar, J.C.; Dolan, C.E.; Mora, M.E.; Nuovo, G.J.; Cole, S.E.; Viapiano, M.S. Fibulin-3 Promotes Glioma Growth and Resistance through a Novel Paracrine Regulation of Notch Signaling. Cancer Res. 2012, 72, 3873–3885.
- Hiddingh, L.; Tannous, B.A.; Teng, J.; Tops, B.B.; Jeuken, J.; Hulleman, E.; Boots-Sprenger, S.H.; Vandertop, W.P.; Noske, D.P.; Kaspers, G.J.; et al. EFEMP1 induces γ-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget 2013, 5, 363–374.
- Zeng, A.-L.; Yan, W.; Liu, Y.-W.; Wang, Z.; Hu, Q.; Nie, E.; Zhou, X.; Li, R.; Wang, X.-F.; Jiang, T.; et al. Tumour exosomes from cells harbouring PTPRZ1–MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 2017, 36, 5369–5381.
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.-G.; Joyce, C.; et al. Mesenchymal Stem Cells Isolated from Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells 2015, 33, 2400–2415.
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; De Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843.
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272.


