 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stefaan W Van Gool | + 6090 word(s) | 6090 | 2021-01-12 07:33:42 | | | |
| 2 | Lily Guo | -4 word(s) | 6086 | 2021-01-22 08:01:03 | | | | |
| 3 | Lily Guo | -4 word(s) | 6082 | 2021-01-24 13:15:37 | | |
Video Upload Options
Immunotherapies represent a promising strategy for glioblastoma multiforme (GBM) treatment. Different immunotherapies include the use of checkpoint inhibitors, adoptive cell therapies such as chimeric antigen receptor (CAR) T cells, and vaccines such as dendritic cell vaccines. Antibodies have also been used as toxin or radioactive particle delivery vehicles to eliminate target cells in the treatment of GBM. Oncolytic viral therapy and other immunogenic cell death-inducing treatments bridge the antitumor strategy with immunization and installation of immune control over the disease. These strategies should be included in the standard treatment protocol for GBM. Some immunotherapies are individualized in terms of the medicinal product, the immune target, and the immune tumor–host contact. Current individualized immunotherapy strategies focus on combinations of approaches. Standardization appears to be impossible in the face of complex controlled trial designs.
1. Introduction
Cancer is the second leading cause of death, accounting for about 1 in 6 human deaths. Worldwide, in 2018, about 9.6 million deaths were due to cancer[1]. Between 2013 and 2017, the cancer death rate (mortality rate) in the US was 158/100,000 individuals per year. The rate of new cases (incidence) in a similar period was 442/100,000 individuals per year[2]. Intensive preclinical and clinical research is being performed to find solutions. In some domains, like pediatric hemato-oncology, major progress has been realized towards a cure through systematic randomized controlled clinical trials (RCTs). In each trial, a new experimental arm is assessed versus the best current treatment as the control arm[3][4], in combination with careful monitoring of (long-term) side effects[5].
Despite being an orphan disease, brain tumors are the leading cause of cancer death in males aged 20 to 39 years and the fourth most common cause of cancer death in females in the same age range[6]. Glioblastoma Multiforme (GBM) is the most frequently diagnosed malignant brain cancer in adults and has the worst prognosis[7][8]. The cause of GBM formation is not known. Ageing that progressively suppresses normal immune surveillance has been mentioned to contribute to GBM cell initiation and/or outgrowth[9]. Irradiation is certainly a cause for GBM formation, and the prognosis of a second malignant GBM is extremely poor[10]. Long-term exposure to higher doses of non-ionising irradiation has been associated with the formation of GBM[11]. Finally, viral infections like CMV (variants) have been mentioned as being potential triggers for GBM formation[12]. The classic pillars of treatment for GBM nowadays are neurosurgery, radiochemotherapy, and maintenance chemotherapy[13][14]. In recent years, the standard of care has not changed. Intensive research in different domains has been performed to improve the prognosis of GBM patients, including research in tumor-treating fields, anti-angiogenic treatments, targeted therapies, oncolytic virus therapy, and immunotherapies. The latter term covers different approaches, like restorative immunotherapy, modulating immunotherapy, passive immunotherapy, adoptive immunotherapy, and active-specific immunotherapy with vaccines [15]. For the development and production of mostly personalized cell-based therapies, Good Manufacturing Practice (GMP) facilities are required.
On the occasion of a regulatory audit in July 2019, in connection to the installation of a new GMP facility at the Immune Oncologic Centre in Köln (IOZK, www.iozk.de) and the running GMP-compatible production of IO-Vac® Dendritic cell (DC) vaccines, a discussion was raised with respect to RCTs within the spectrum of delivered individualized multimodal immunotherapy (IMI) activities. The background of this question is a subject of current global debate regarding the future use of controlled RCTs to obtain evidence of the efficacy of immunotherapies. This was exemplified by the symposium organized in Brussels on 22 April 2020, entitled “are randomized trials obsolete?” [16]. On 27 May 2015, the IOZK received a certificate of GMP manufacturing compliance (DE_NW_04_GMP_2015_0030) and approval to produce specific autologous anti-tumor DC vaccines for intradermal injection (DE_NW_04_MIA_2015_0033). Since then, multimodal immunotherapy has been implemented in several domains of cancer. Both certificates were renewed on 20 May 2020 (i.e., DE-NW-04-GMP-2020-0054 and DE-NW-04-MIA-2020-0017).
The IOZK is a translational immune-oncology center specializing in the fast translation of emerging novel insights derived from multiple domains of immunotherapy into clinical applications for use on a compassionate basis (“Individueller Heilversuch”) for patients with cancer. The key medicinal product is IO-Vac®, which is an approved Advanced Therapy Medicinal Product (ATMP). The IO-Vac® vaccine consists of autologous mature DCs loaded with autologous tumor antigens and matured with danger signals including the Newcastle Disease Virus (NDV). Over the years, the IOZK has established its value for the treatment of cancer patients. Several case reports and retrospective analyses of patient groups have been published[17][18][19][20]. For the current retrospective analysis, the database was fixed at 28/06/2020, including all records registered from 01/06/2015 to 31/05/2020. Over this 5 year time period, 1456 medical records were initiated at the IOZK. The patients came from 69 countries (with 46% from Germany). From this group of patients, 1098 patients agreed to go through an immune-oncologic evaluation and immunodiagnostic blood sampling in order to study the cell numbers and functioning of their immune compartment, the tumor–host immune interactions, their general health status, and their infection status. Ultimately, 651 were able to, and individually consented to, starting multimodal immunotherapy, after being extensively informed about all aspects of the treatment. These patients belonged to all categories of cancer disease. The three most frequent cancer disease categories were neuro-oncology (42%), digestive oncology (18%), and breast cancer (11%). The domain of neuro-oncology represents almost half of all patients effectively treated at the IOZK. This group of patients is still a very heterogeneous group, including multiple types of brain cancer disease, and including patients at different stages of disease. From the 276 patients, 171 patients (62%) were recorded as having GBM.
2. Current anti-cancer treatment strategies for GBM
GBM is one of the leading causes of death due to cancer in humans and is a major burden for the community[21][22]. Earlier, in the period of poor imaging possibilities, neurosurgery was the only treatment for GBM, and this was mostly performed to make pathological diagnoses and to temporarily relieve symptoms. Only during the last century other treatment modalities became available, of which radiotherapy was the first approach. Radiotherapy became part of the standard care for GBM in the 1940s. The authors could not find any RCTs from that period. Evidence of the efficacy of radiotherapy was certainly created by the demonstration of a dose–response relationship [23]. An RCT, including radiotherapy, chemotherapy, or best available care for anaplastic astrocytoma, was realized around the same period, clearly demonstrating the effect of radiotherapy on OS[24]. Radiotherapy was shown to improve the median OS of GBM patients by some months, and 60 Gy appeared to be the most efficacious and safe dose[25]. More recently, chemotherapeutic agents and combination treatments have been implemented. The addition of temozolomide (TMZ) during radiotherapy, followed by TMZ maintenance chemotherapy (TMZm) further improved the median OS (again, by some months). Proof of evidence was generated through a prospective RCT. In this trial, patients were stratified by World Health Organization (WHO) performance status, type of surgery, and institution[13]. Later on, the data were presented according to an adapted Recursive Partitioning Analysis (RPA) classification, which included age, WHO performance status, extent of surgery, and mental status as variables, resulting in Class III, Class IV, and Class V patients[14].
The MGMT promotor methylation status of the tumor was rapidly recognized as a key factor in the efficacy of TMZ[26]. Retrospective analyses of available MGMT promotor methylation data in relation to the survival data in the RCT have shed new light on such data[27]. The progression-free survival (PFS) benefit attained through the addition of TMZ to surgery and radiotherapy lost its significance in MGMT promotor unmethylated patients, and the gain in median OS was only 0.8 months (i.e., 24 to 25 days), albeit still being significant. Although the results of the original prospective RCT were published 15 years ago, this so-called Stupp regimen is still the standard of care world-wide for both MGMT promotor-methylated and unmethylated patients. Due to its major effect, even in multivariate analyses, the MGMT promotor methylation status has become part of the in/exclusion criteria for RCTs or is used in the stratification of the randomization. Step by step, more insight into the transcriptomic and genomic dimensions were found, and GBM molecular stratification appeared, pointing to EGFR, NF1, and PDGFRA/IDH1 as playing roles in triggering intracellular pathways but also in influencing the response to anti-GBM treatments, formation of the tumor microenvironment, and tumor spread[28]. A further molecular biological analysis including epigenetic profiling unraveled at least six sub-types of GBM, all having different disease characteristics and prognoses[29].
Radiotherapy and chemotherapy treatment modalities are directed against the cancer itself, but cause acute and long-term side effects to the body. These treatments might, however, also have important effects on the immune system. It is already well-known that neurosurgical removal of the tumor transiently “relieves” the systemic immune system from tumor-induced immunosuppression[30], which returns upon disease progression [31]. Both radiotherapy and TMZ have effects on inflammation and the systemic immune compartment but may also influence the tumor microenvironment[32][33][34][35][36].
The improvement in therapeutic approaches has been paralleled by improvements in imaging technologies. Furthermore, knowledge of the molecular biology domain has increased rapidly, thereby introducing new approaches to the development of targeted treatments with less toxicity. A whole series of targeted therapies have been investigated for GBM, as single drugs in relapsed patients or as add-ons to the Stupp regimen[37][38][39][40][41][42]. Fast molecular biological diagnostic procedures and initiation of adapted drug combinations have opened the door to so-called personalized medicine. For the first time, a particular tumor entity is no longer treated according to pre-designed treatments or study protocols but may become adapted to the individual tumor biology profile of each individual patient. Along the same line, targeted therapies can be implemented to treat any type of cancer, regardless of where in the body it started or the type of tissue from which it developed, pointing to the term “tumor-agnostic treatment”[43].
3. Immunotherapy for GBM
Immunotherapy was called a “break-through for cancer” by Science in 2013. In 2011 and 2018, the Nobel Prize for Medicine was devoted to researchers who discovered important insights into the use of immunotherapy for cancer. At present, immunotherapy is definitively an important pillar in anticancer treatment in general, including for GBM. Immunotherapies generally do not point to the molecular biological characteristics inside tumor cells, but rather, to the surface of tumor cells, including a heterogeneous number of known and unknown tumor antigens[44] and immune costimulatory and inhibitory molecules on the surface[45] [46][47], as well as the production of cytokines and chemokines that influence the immune system and the inflammatory response[48]. Terms like “cold” versus “hot” tumors, describing differences in lymphocyte infiltration in the tumor microenvironment, have become highly relevant[49]. The tumor mutational burden might be related to the presence of more or less tumor antigens on the surface[50][51]. Some genetic abnormalities, such as the p53 mutation, affect the extent of the expression of MHC molecules on the tumor cell surface[52]. An immunogram is created using the tumor foreignness, general immune status, immune cell infiltration, absence/presence of checkpoints, absence/presence of soluble inhibitors, absence/presence of inhibitory tumor metabolism, and sensitivity of the tumor to immune effectors[53]. The recognition that almost half of GBM tumors consist of cells belonging to the myeloid compartment (e.g., M1/M2 macrophages, tumor-associated macrophages, myeloid-derived suppressor cells, microglia) makes the understanding of the response to treatment and the ultimate outcome of patients more difficult [54][55]. Overall, the tumor microenvironment can be categorized into three functional sub-types that play significant roles in the outcomes of patients[56]. Interestingly, Hallaert et al.[57] also observed that the connection of GBM to the sub-ventricular zone appears to be a novel, independent risk factor leading to a worse prognosis. An association between sub-ventricular zone contact and markers related to the epithelial–mesenchymal transition was discovered[58]. Moreover, there is a definite influence of the tumor and the response to treatment on the systemic immune and inflammatory compartments[59]. Finally, steroids, radiotherapy, and chemotherapy all influence antitumor immune responses [36]; this, again, differs from patient to patient. A strong correlation was found between OS and the systemic immune profile after neurosurgical resection and after radiochemotherapy when the extent of resection and timing of immunotherapy were taken into account [60].
Immunotherapy covers many treatment modalities. Some of these treatment modalities, such as cytokines, are not personalized to each patient. Other drugs, like antibodies, are themselves not personalized, but are directed against personalized targets on the tumor cell surface. Immunomodulation with checkpoint inhibitors is considered to belong to the domain of tumor-agnostic treatments[43]. For adoptive cell therapies with chimeric antigen receptor (CAR) T-cells or T cell receptor-transduced T-cells, both the ATMP itself and the target are personalized[61][62][63]. In the domain of active specific immunotherapy with vaccines, the vaccine itself can be an individual product at the level of the antigen carrier (like autologous DCs) and/or at the level of the antigen, such as tumor-derived antigens and, especially, highly individualized tumor-specific epitopes [64][65][66]. Extensive individualization of the treatment drug and target makes the design of a homogeneous experimental arm versus control arm very challenging. By introducing immunotherapy into a first-line treatment combination, new dimensions of personalization for the treatment of GBM have become clear.
Overwhelming evidence of the efficacy of DC vaccination for GBM has been presented in preclinical models [64-76[67][68][69][70][71][72][73][74][75][76][77][78][79], as well as in phase I and early phase II non-controlled clinical trials[80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144]. The observation of long-term OS in patients after DC vaccination, noting that the intervention abruptly changes the 100% lethality of disease, has generated strong evidence of its efficacy. Several meta-analyses have demonstrated a significant improvement in long-term OS when GBM patients were treated with DC vaccines as compared with patients given a control treatment[145][146][147][148][149]. Of note, the latter meta-analysis also demonstrated a lack of additional toxicity due to immunotherapy in comparison with the standard of care. This means that the set-up of new phase I or phase IIa clinical trials is no longer innovative.
As has already been pointed out for the changes in molecular sub-clones over time during treatment, evidence that tumor–host immune interactions can also change over time has been obtained. Examples are the downregulation of specific target antigens[150] or the upregulation of PDL1 on tumor cells, allowing them to escape from immune attack during immunotherapy[151]. Dynamic changes in the expression of tumor-associated antigens and immunosuppressive factors in the tumor microenvironment during treatment have also been described[144].
4. Individualized Multimodal Immunotherapy for GBM
Two conclusions have become clear: 1) Individualized treatment at multiple levels, with strategies that follow the dynamic changes in both the tumor and host (e.g., immunity, inflammation) during treatment, is needed for the treatment of GBM; and 2) IMI is becoming part of the first-line treatment for GBM, integrated into the standard of care. The potential inherent immunization component of standard anti-GBM strategies (e.g., neurosurgery, radiotherapy, and chemotherapy) and the proven induction of immunogenic cell death (ICD) through the use of innovative anti-cancer treatments (e.g., modulated electrohyperthermia, oncolytic virus therapy)[152][153] should be exploited and strengthened with active immunization strategies and further optimized with immunomodulatory strategies in the context of optimized complementary medicine. Only broad and long-term immune control over GBM is able to induce long-term OS. The rationale of such a strategy has been published[154]. In sections 8 to 10, we aim to illustrate several elements discussed in the former narrative review with observational data obtained by a retrospective analysis of our own patient records. Over the years, we have developed a multistep treatment approach, in which ICD is induced by the combination of bolus injections with NDV and modulated electrohyperthermia (mEHT) integrated as an add-in treatment during the alkylating mode of tumor cell killing induced by TMZm chemotherapy, followed by IO-Vac® DC vaccination, in order to actively induce an antitumor immune response in combination with individually adapted immunomodulatory strategies. Finally, ICD treatment is maintained with immunomodulation[151].
Of the 171 recorded patients with GBM in the IOZK database, 90 patients (53%) received IMI in combination with first-line treatment for compassionate use (“individueller Heilversuch”). For the current retrospective analysis, we excluded all patients below 18 and above 75 years of age, patients with a proven pre-history of low-grade glioma and/or proven IDH1 mutation, patients with proven histone mutations, and patients with radiotherapy-induced GBM as a second malignancy, leaving 70 patients for analysis.
The final patient group consisted of 70 patients (33 females, 37 males). The median age of the patients was 50 years (range 18–69 years). The median Karnofsky performance index score was 90 (range 50–100). Clinical patient characteristics are shown in Figure 1. The patients presented for immune diagnostic blood sampling at IOZK at a median of 3 months after operation (range 0.5–23 months); hence, most sampling was done after radiochemotherapy. General blood counts and immune cell counts and functioning, as analyzed in the routine clinical laboratory, are shown in Figure 2A. A large proportion of patients were lymphopenic at that time—caused by the radiochemotherapy—a finding compatible with published data[36].
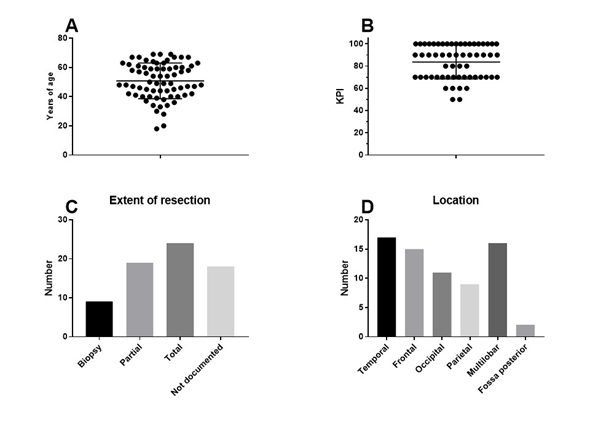
Figure 1. Clinical characteristics of the patients. Seventy adults with primary GBM receiving first-line standard of care in combination with individualized multimodal immunotherapy were included in this retrospective analysis: (a) age distribution; (b) distribution of Karnofsky performance index scores; (c) reported resection (number of patients); and (d) reported location (number of patients).
The MGMT promotor methylation status of the resected tumor was documented in 52 cases, from which 32 patients were categorized as unmethylated. In 67 patients, the presence of circulating tumor cells (CTCs) was investigated at the time of immune diagnostic blood sampling. Blood samples were sent to Biofocus (https://www.biofocus.de/) for analysis. In 28 patients, no CTCs were detected. CTCs were detected in 39 patients, based on the presence of large cells with mRNA expression for specific oncogenes (EGFR, ERBB2, C-kit, and Telomerase) above a cut-off in comparison with house-keeping mRNA expression. In nine patients, mRNA expression for PDL1 was above the cut-off value of 2 (Figure 2B). In 11 patients, increased mRNA expression for MGMT was observed, of which seven patients were histologically classified as being MGMT promotor unmethylated, while three patients were classified as being methylated (one patient had an unknown histology result). For 16 patients who had CTC without increased mRNA for MGMT, six samples matched with a histologic classification as MGMT promotor methylated, while nine samples mismatched with the histologic classification and were classified as MGMT promotor unmethylated. It should be noted that the results for both histology-based MGMT promotor methylation and the mRNA expression of MGMT in CTCs were derived from continuous variables linked with methylated versus unmethylated status. Moreover, GBM might depict heterogeneity for MGMT promotor methylation, such that histology sampling errors cannot be excluded. Finally, the presence of CTCs was investigated at a median of 3 months after operation, thereby potentially illustrating clonal evolution during the first period of radiochemotherapy.
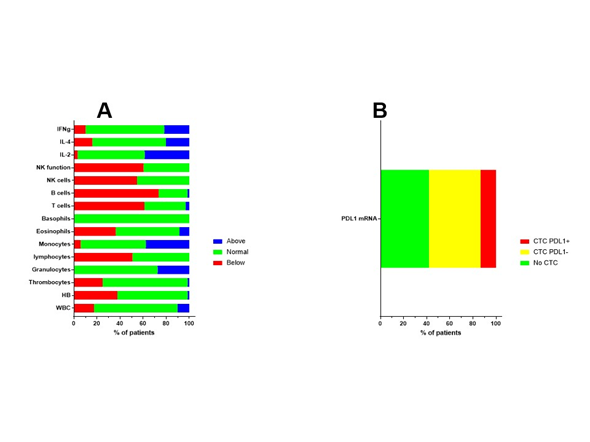
Figure 2. Immune variables before the start of immunotherapy. (a) Before the start of individualized multimodal immunotherapy, blood was drawn and sent to the routine clinical lab for analysis. Different immune variables are shown. The percentages of patients with values above (blue), within (normal, green), or below (red) the normal range are shown. (b) Blood was also sent to Biofocus in order to detect circulating tumor cells based on mRNA expression of GBM-related oncogenes. When cells were detected, the RNA expression for PDL1 was subsequently analyzed and cells were defined as negative (yellow) or positive (red) for the expression of mRNA for PDL1.
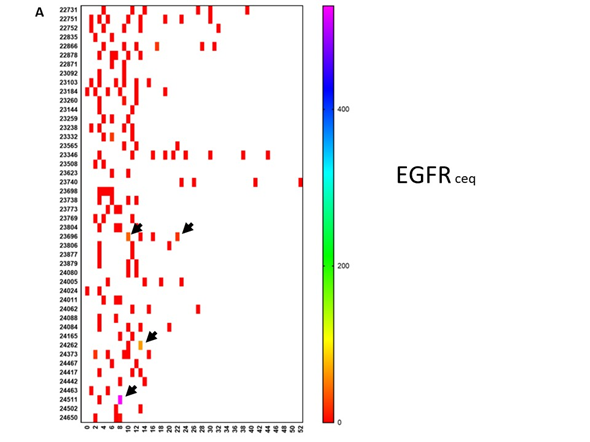
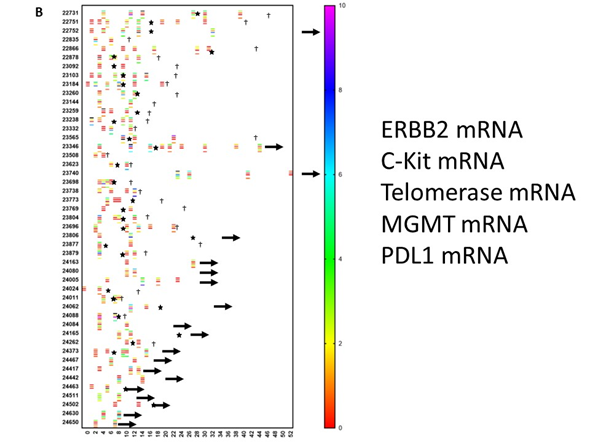
Figure 3. Evolution of oncogene expression in circulating tumor cells. Circulating tumor cells were measured repetitively in 46 patients. Each patient is referenced with a number on the Y-axis. Time in months is indicated on the X-axis. The scale of the color of each test is shown on the right-hand side. (a) The level of mRNA expression for EGFR is expressed as the ceq (cell equivalent). Most of the values are negative (red). However, as indicated by the arrows, some patients showed an upregulation of EGFR during the disease course. (b) This shows a similar data set-up as in panel A. For each patient, data on mRNA expression are relative to the house-keeping gene GADPH and are shown in a particular color, for which the scale is shown on the right-hand side. For each patient, up to 5 lines are shown at different moments during the disease course. From top to bottom, values for ERBB2, c-Kit, telomerase, MGMT, and PDL1 are shown for each patient. Stars indicate new events. A † indicates when the patient died. An arrow to the right indicates the time at which the patient was censored in the analysis.
To further illustrate the biological evolutions that occurred during treatment, we looked at the evolution of mRNA expressions in CTCs for the oncogenes EGFR, ERBB2, C-kit, and Telomerase, as well as the mRNA for MGMT and PDL1 over time. For this, available data on mRNA expression in CTCs were sampled and put on a time line for each patient starting from neurosurgery. As mentioned earlier, the patients started immunotherapy at different time points and were treated individually. This is reflected by the use of an individual monitoring schedule for each patient. Figure 3A shows the evolution of the EGFR mRNA expression over time for individual patients. Changes in mRNA expression were only observed in three patients. On the contrary, as shown in Figure 3B, mRNA expression for ERBB2, C-kit, Telomerase, MGMT, and PDL1, in comparison to mRNA expression for the house-keeping gene NADPH, clearly changed over time and was often visible around the appearance of relapse. As an example, the mRNA expression for PDL1 in the CTCs of patients 22731 and 23346 was low at start of immunotherapy treatment. These values, however, increased during treatment and were ultimately accompanied by relapse. Patient 23346 was treated with pembrolizumab. Patient 24005 also had a very high level of mRNA expression for PDL1 when the CTCs became positive for the first time. This patient also received four doses of pembrolizumab, after which the CTCs became negative for the PDL1 marker. These descriptive data indirectly demonstrate the possible relative changes in sub-clones during the disease process, which might contribute to the occurrence of relapse and which might be monitored for timely intervention. GBM is thus a dynamic tumoral process. The descriptive analysis illustrates the heterogeneity within these 70 patients at the levels of clinical risk factors, immune variables in the peripheral blood, and molecular tumor biology data, as well as the dynamic processes occurring in the molecular biology of the tumor.
The patients received IMI within the first-line treatment. Table 4 shows the details of the IMI treatment. A median of two (range 0–5) IO-Vac® DC vaccines, 25 (range 0–77) NDV administrations, and 30 (range 0–77) sessions of modulated electrohyperthermia were given to each patient. One IO-Vac® DC vaccine consisted of a median of 12.2 x 106 autologous mature IO-Vac® DCs loaded with autologous tumor proteins derived from tumor lysate (when available and appropriate to the GMP requirements) or obtained as ICD-therapy-induced, serum-derived, antigenic, extracellular microvesicles and apoptotic bodies.
Table 3. Treatment details.
|
|
Vaccine |
Local Hyperthermia |
NDV |
DC total |
DC/vaccine |
|
N |
69 |
70 |
69 |
70 |
112 |
|
Minimum |
0 |
0 |
0 |
0 |
2 400 000 |
|
25%P |
1 |
13 |
17 |
6 950 000 |
8 075 000 |
|
Median |
2 |
25 |
30 |
20 115 000 |
12 200 000 |
|
75% |
2 |
42 |
42 |
34 200 000 |
19 145 000 |
NDV: Newcastle Disease Virus.
5. A case of complete remission and specific T-cell response
In our retrospective analysis of these 70 patients, one particular innovative finding emerged. As the tumor-specific antigens were not known when using tumor lysate or serum-derived, extracellular vesicles, immune monitoring in our clinical setting was not a major focus. Nevertheless, proof of principle of ICD therapy in combination with TMZ maintenance chemotherapy followed by vaccination with IO-Vac® DC vaccines loaded with ICD therapy-induced, serum-derived, antigenic, extracellular microvesicles and apoptotic bodies[151] for the induction of a tumor-neo-epitope-specific immune response was demonstrated in one patient. This 18-year-old patient had an incomplete resection of a left frontal lobe IDH1 wild-type and MGMT unmethylated GBM. Their tumor mutational burden was low (0.5 variants/megabase), and there was no evidence for microsatellite instability or germline variants. She was treated with radiochemotherapy and, subsequently, five cycles of TMZm chemotherapy (Figure 4). At presentation, her Karnofsky performance index was 90. She was lymphopenic with 279/ul CD3+ T-cells, 78/ul CD19+ B-cells, and 56/ul CD16+CD56+ NK cells. Her NK cell functioning was below the reference value. She had Th2/Th17 skewing. Her CTCs showed upregulated expression (3.44) of mRNA for MGMT, which was compatible with the known unmethylated MGMT promotor status. The expression of mRNA for PDL1 was 1.13 (cut-off for positivity = 2). She continued treatment for another seven TMZm chemotherapy cycles combined with 5-day ICD treatments, which were given during each TMZm cycle at days 8 to 12. Afterwards, she received two IO-Vac® DC vaccinations loaded with ICD treatment-induced, serum-derived, antigenic, extracellular microvesicles and apoptotic bodies. Later on, she received two IO-Vac® DC vaccines loaded with tumor-specific peptides based on the individualized tumor-specific neo-antigen detection tests performed at CeGaT (www.CeGaT.de). At the time of writing, she receives monthly maintenance ICD treatments and is still in complete remission. Interestingly, we were able to monitor the tumor antigen-specific T-cell responses as, in this case, the neo-antigens were known. A first sample was available after the fifth TMZm cycle, prior to the addition of multimodal immunotherapy. A second sample was available at the time of blood sampling to prepare the second IO-Vac® DC vaccine (Figure 4). From the data, it is clear that surgery, radiochemotherapy, and five cycles of TMZm did not induce a tumor-specific T-cell response. However, the addition of seven ICD treatments to the last seven TMZm treatments and the first IO-Vac® DC vaccine loaded with ICD-treatment-induced, serum-derived, antigenic, extracellular microvesicles and apoptotic bodies generated a clear tumor antigen-specific CD4+ and CD8+ T-cell response. The impact of this observation might be meaningful. First, it is not mandatory to have fresh frozen tumor material to prepare a tumor lysate as an antigenic source. This avoids the critical challenges of yielding, freezing, transporting, analyzing, and preparing tumor material within a GMP context. Secondly, and even more importantly, if molecular subclones can change over time, their antigenic profiles can change as well. ICD treatment allows the yield of tumor antigens that are expressed within the body at the time of treatment and makes it possible for the vaccine to immunize against the antigens that are actually present, instead of tumor antigens identified at the time of tumor resection prior to radiotherapy and chemotherapy and hence prior to eventual treatment-induced tumor clone changes.
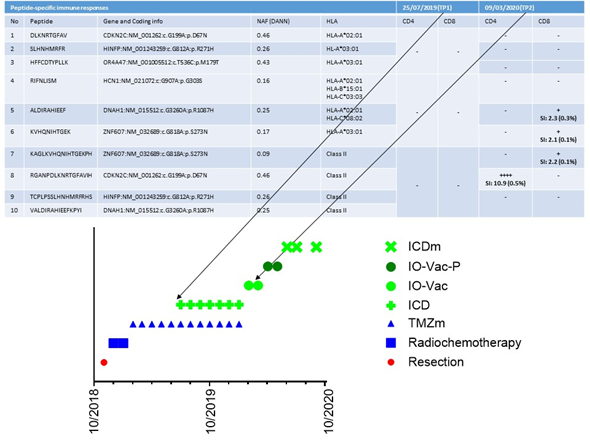
Figure 4. Detection of tumor-specific T-cell clones. The treatment timeline for patient 24442 and its multiple components are shown in the bottom part of the figure. TMZm indicates 5 days of temozolomide maintenance treatment in cycles (every 28 days). ICD indicates immunogenic cell death (ICD) treatment consisting of the combination of five injections with Newcastle Disease Virus and 5 sessions of modulated electrohyperthermia. IO-Vac indicates a vaccination cycle including six ICD treatments and an injection of IO-Vac® DC vaccine, consisting of autologous mature dendritic cells loaded with ICD-treatment-induced, serum-derived, antigenic, extracellular microvesicles and apoptotic bodies. IO-Vac-P vaccination cycles are equal to IO-Vac vaccination cycles, but the DCs are loaded with tumor-specific neo-peptides. The upper part of the curve shows the specific peptide sequences, the respective gene and coding information, the Novel Allele Frequency (NAF), and the HLA phenotype. TP1 and TP2 indicate the two respective time points at which T-cells were frozen for immune monitoring purposes. SI is the stimulation index, the ratio of polyfunctional activated CD4+ or CD8+ T-cells (positive for at least two activation markers from CD154, IFN-g, TNF, and/or IL-2) in the peptide-stimulated sample, compared with the unstimulated control. Additionally, the percentage of activated CD4+ or CD8+ T-cells (positive for at least one activation marker of CD154, IFN-g, TNF, and/or IL2) above the background and after in vitro amplification is given. This percentage does not directly reflect the frequencies in vivo. SI > 2: weak response (+); SI > 3: positive response (++); SI > 5: strong response (+++); SI > 10: very strong response (++++). Peptides 1–3, 4–6, and 7–10 were pooled for the analysis of TP1. Peptides 9 and 10 were pooled for the analysis of TP2.
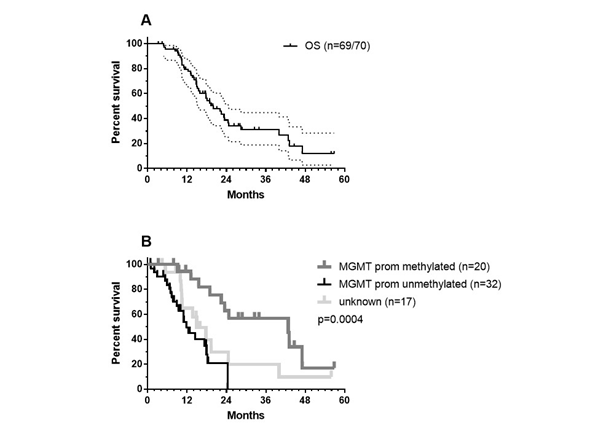
Figure 5. OS data: (a) OS data of the total patient group (CI95% values are shown). One patient out of 70 was lost during the follow up period and (b) the patient group was divided according to MGMT promotor methylation status—methylated (grey), unmethylated (black), data not registered (light grey). The p-values show significance using the Log-rank (Mantel–Cox) test.
6. Results in term of OS
As we did not have reference radiology for the independent assessment of new events, given the need to follow the iRANO criteria[155][156] and as OS is certainly the most important outcome in the context of immunotherapy, we focused on the OS of the patients. As described earlier in section 8, the retrospectively analyzed group of 70 patients was a heterogeneous group of patients. They best reflected adapted RPA class 4 patients, as published by Stupp et al.[27]. This reference was used as a historical control. Data were analyzed using GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla California, USA, www.graphpad.com). One patient was lost during follow up. The median OS was 20.03 months (Figure 5A) with a 2-year OS of 38.83% (CI95%: +13.04, -13.25) and a 3-year OS of 31.41% (CI95%: +13.35, -12.55). Patients younger than 50 years (21 MGMT promoter unmethylated, 7 methylated, and 7 unknown) had a median OS of 22.07 months, which was not statistically different from the median OS of 18.07 months observed for patients older than 50 years (11 unmethylated, 13 methylated, and 11 unknown). We found MGMT promotor methylation status to be significant factor (Figure 5B, log-rank test: p = 0.0004): patients (n = 20) with patients with MGMT promotor methylation having a median OS of 42.85 months versus 11.77 months for patients with unmethylated MGMT promotor status (n = 32).
For 16 patients, local therapy (neurosurgery, radiochemotherapy) was followed by IMI (Group 1). ICD therapy (the combination of IV bolus injections of NDV together with mEHT[151]) integrated into the TMZm chemotherapy cycles followed by IO-Vac® DC vaccines and maintenance ICD therapy) was given to 46 patients (Group 2). Eight patients started with IMI after the last TMZ maintenance chemotherapy cycle (Group 3). The OS data were significantly different for the three treatment groups: 13.08 months for group 1, 22.46 months for group 2, and undefined for group 3 (median follow-up of surviving patients: 28.59 months, range 26.3–55.9 months; Figure 6A). The latter likely points to favorable selection of a small group of patients who experienced no events until after the end of the maintenance chemotherapy and profited from subsequent IMI treatment. The median OS of the patients with unmethylated MGMT promotor status was 11.25 months for group 1 (n = 7) and 18.07 months for group 2 (n = 21), with a 2-year OS of 0% in group 1 versus 17.18% (CI95% +31.65, -15.86) in group 2 (Figure 6B, log-rank test: p = 0.0273). The median OS of the patients with methylated MGMT promotor status was 42.85 months for group 2 (n = 15) with a 2-year OS of 66.66% (CI95%: +19.3, -32.96). There were only three patients in group 1 who had a tumor with MGMT promotor methylation status. Within treatment group 2, the differences in the OS curve due to MGMT promotor methylation status was significant (Figure 6C, Log-rank test: p = 0.0036).
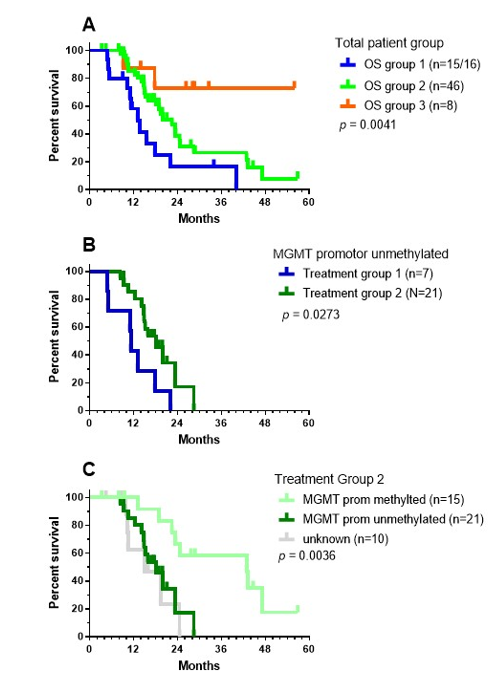
Figure 6. OS data for the treatment groups. As explained in the text, patients were categorized into three different treatment groups: (a) OS data for the three different patient groups; (b) OS data for the patients without MGMT promotor methylation belonging to treatment groups 1 (blue) and 2 (green); and (c) OS data of patients from treatment group 2, divided according to MGMT promotor methylation status. The p-values show the significance calculated using the Log-rank (Mantel–Cox) test.
Considering these data and the data published by Stupp et al.[27], patients with MGMT promotor unmethylated status have no relevant benefit from the addition of TMZ[27] or from treatment with IMI alone. However, the data from treatment group 2 suggest the potential benefit of the integration of ICD treatment within TMZm chemotherapy followed by IO-Vac® DC vaccinations, which was found to increase the median OS by about 6 months. On the other hand, treatment with IMI after local therapy for patients with MGMT promotor methylated GBM yielded similar median OS data as treatment with TMZm chemotherapy, while the integration of ICD treatment with TMZm chemotherapy followed by IO-Vac® DC vaccinations increased the median OS further—by about 18 months. These data are worth validating in prospective clinical research.
7. Conclusions
In this paper, we reviewed discuss treatment modalities for GBM and focus on the role of immunotherapy. We illustrated these challenges in a retrospective analysis of GBM patients treated at IOZK. GBM treatment is affected by the complexity of the tumor, the complexity of the immune and inflammatory micro-environment, the complexity of combined treatment approaches, and the complexity of dynamic changes occurring within the tumor, the tumor microenvironment, and the immune system. The set-up of immunotherapy RCTs for GBM is affected by the need for multiple stratifications to create an appropriate control group. Both stratification and the read-out of OS necessitate the use of a large number of study participants. The costs to run such RCTs are related not only to the Good Clinical Practice (GCP) documentation but also to the production of the ATMP in a GMP environment. As already mentioned, there has been an abundance of smaller phase I and phase II single-arm clinical trials, and several meta-analyses have shown the efficacy and safety of DC vaccination for GBM treatment. The high cost of non-innovative small RCTs with predicted negative outcomes due to a lack of appropriate stratification makes them not appropriate at this time due to limited resources. Models for OS and immunotherapy responses in patients with GBM based on DNA-methylation-driven, gene-based, molecular classifications and multi-omic analyses may be better tools for predicting the efficacy individually for each patient[157]. However, patients suffering today from GBM need better treatments, including the use of first-line immunotherapy to fight their cancers.
The multimodality of immunotherapy, besides the standard of care, presents a further challenge to the classic step-wise research methodology used in clinical research. IOZK aims to contribute to the general knowledge and communicate their gained experiences to the wider scientific community. The ATMP IO-Vac® DC vaccine has been used as part of IMI treatment in the framework of “Individueller Heilversuch”. Each patient is treated at all personalized levels necessary. For each individual patient, the best possible solution for their medical needs is worked out. Prospective medical research questions on groups of patients are not generated. Nevertheless, the IOZK aims to repetitively freeze the database at certain time points and sample patient data to carry out scientifically correct retrospective analyses. The current analysis suggests that the addition of ICD treatment with NDV and mEHT during the application of TMZ maintenance chemotherapy, followed by IO-Vac® DC vaccinations and maintenance ICD treatment, may be beneficial for both MGMT promotor unmethylated and methylated GBM patients.
A final challenge in the broad implementation of innovative therapies like IMI, given that its effectiveness has been accepted without the use of RCTs, is the expected cost versus the length of additional survival. The production of personalized ATMPs under GMP conditions is expensive. The control over the ATMP quality, and hence over the costs, belongs to the authorities, who are also responsible for health policies in general. This topic points to the need for a cost–effectiveness analysis to compare the costs and outcomes of treatment options. The aim is to ensure the greatest possible health benefits are attained with a given budget. For macroeconomic considerations, cost-effectiveness thresholds (CETs) are usually related to the gross domestic product (GDP) per capita [158]. To the best of our knowledge, no representative data related to the integration of IMI into standard of care treatment of GBM are available. Single studies have appraised the additional cost of targeted therapies per year of survival if combined with chemotherapy. In non-small-cell lung cancer, for example, the additional cost for using atezolizumab in combination with carboplatin/nab-paclitaxel amounts to 333,199 USD per quality-adjusted life year[159]. Understandably, the respective costs are difficult to calculate and depend heavily on the type of cancer, the treatment in question, and the parameters used in the cost-effectiveness study. Accordingly, different methods have been described[160]. What we can say is that overall spending on cancer drugs has been increasing. For example, spending on cancer drugs rose from €7.6 billion in 2005 to €19.1 billion in 2014 in the EU[161]. For some types of cancer, calculations are available. Between 2007 and 2012, the mean amounts of money spent in the first year after diagnosis was $35,849, $26,295, $55,597, and $63,063 for breast, prostate, lung, and colorectal cancers, respectively[162]. In view of the overall increase in costs, even higher expenditure must be expected today. A more recent study on breast cancer reported the average cost per patient in the year after diagnosis as being between $60,637 and $134,682, depending on the cancer stage [163]. Newly approved pharmaceuticals easily cost more than $100,000 US per year. Costs are driven by various factors that are not proportional to their often modest additional benefits[164][165], exceeding the cost-effectiveness thresholds. In comparison, the addition of modulated electrohyperthermia to dose-dense temozolomide for the treatment of recurrent GBM has proven to be cost-effective[166]. Taken together, it is methodically difficult to compare the cost–benefit ratio of the integration of IMI into standard of care treatment. Considering the far higher overall costs of targeted therapies, one may expect that the ratio would not be unfavorable.
References
- Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 18 December 2020).
- Available online: https://www.cancer.gov (accessed on 18 December 2020).
- Creutzig, U.; Zimmermann, M.; Dworzak, M.N.; Ritter, J.; Schellong, G.; Reinhardt, D. Development of a curative treatment within the AML-BFM studies. Klin. Padiatr. 2013, 225 (Suppl. 1), S79–S86, doi:10.1055/s-0033-1337968.
- Schrappe, M.; Reiter, A.; Ludwig, W.D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: Results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 2000, 95, 3310–3322.
- Loning, L.; Zimmermann, M.; Reiter, A.; Kaatsch, P.; Henze, G.; Riehm, H.; Schrappe, M. Secondary neoplasms subsequent to Berlin-Frankfurt-Munster therapy of acute lymphoblastic leukemia in childhood: Significantly lower risk without cranial radiotherapy. Blood 2000, 95, 2770–2775.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. Ca Cancer J. Clin. 2020, 70, 7–30.
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemi-ologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996, doi:10.1158/1055-9965.EPI-14-0275.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820, doi:10.1007/s00401-016-1545-1.
- Ladomersky, E.; Scholtens, D.M.; Kocherginsky, M.; Hibler, E.A.; Bartom, E.T.; Otto-Meyer, S.; Zhai, L.; Lauing, K.L.; Choi, J.; Sosman, J.A.; et al. The Coincidence Between Increasing Age, Immunosuppression, and the Incidence of Patients with Glioblastoma. Front. Pharm. 2019, 10, 200, doi:10.3389/fphar.2019.00200.
- Izycka-Swieszewska, E.; Bien, E.; Stefanowicz, J.; Szurowska, E.; Szutowicz-Zielinska, E.; Koczkowska, M.; Sigorski, D.; Kloc, W.; Rogowski, W.; Adamkiewicz-Drozynska, E. Malignant Gliomas as Second Neoplasms in Pediatric Cancer Survivors: Neuropathological Study. Biomed. Res. Int. 2018, 2018, 4596812, doi:10.1155/2018/4596812.
- Hardell, L.; Carlberg, M.; Hansson Mild, K. Use of mobile phones and cordless phones is associated with increased risk for glioma and acoustic neuroma. Pathophysiology 2013, 20, 85–110, doi:10.1016/j.pathophys.2012.11.001.
- Soderberg-Naucler, C.; Johnsen, J.I. Cytomegalovirus infection in brain tumors: A potential new target for therapy? Oncoimmunology 2012, 1, 739–740, doi:10.4161/onci.19441.
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoom, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Eng. J. Med. 2005, 352, 987–996.
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the Eortc-Ncic trial. Lancet Oncol. 2009, 10, 459–466.
- De Vleeschouwer, S.; Van Gool, S.W.; Van Calenbergh, F. Immunotherapy for malignant gliomas: Emphasis on strategies of active specific immunotherapy using autologous dendritic cells. Childs Nerv. Syst. 2005, 21, 7–18.
- Available online: https://www.iddi.com/news/news-events/symposium-are-randomized-trials-still-needed-in-2020/ (accessed on 18 December 2020).
- Schirrmacher, V.; Bihari, A.S.; Stucker, W.; Sprenger, T. Long-term remission of prostate cancer with extensive bone metastases upon immuno- and virotherapy: A case report. Oncol. Lett. 2014, 8, 2403–2406.
- Schirrmacher, V.; Stucker, W.; Lulei, M.; Bihari, A.S.; Sprenger, T. Long-term survival of a breast cancer patient with extensive liver metastases upon immune and virotherapy: A case report. Immunotherapy 2015, 7, 855–860, doi:10.2217/imt.15.48.
- Van Gool, S.W.; Makalowski, J.; Feyen, O.; Prix, L.; Schirrmacher, V.; Stuecker, W. The induction of immunogenic cell death (ICD) during maintenance chemotherapy and subsequent multimodal immunotherapy for glioblastoma (GBM). Austin Oncol. Case Rep. 2018, 3, 1010.
- Van Gool, S.W.; Makalowski, J.; Bonner, E.R.; Feyen, O.; Domogalla, M.P.; Prix, L.; Schirrmacher, V.; Nazarian, J.; Stuecker, W. Addition of Multimodal Immunotherapy to Combination Treatment Strategies for Children with DIPG: A Single Institution Experience. Medicines 2020, 7 29, doi:10.3390/medicines7050029.
- Cao, J.X.; Zhang, X.Y.; Liu, J.L.; Li, D.; Li, J.L.; Liu, Y.S.; Wang, M.; Xu, B.L.; Wang, H.B.; Wang, Z.X. Clinical efficacy of tumor antigen-pulsed DC treatment for high-grade glioma patients: Evidence from a meta-analysis. PLoS ONE 2014, 9, e107173.
- Wang, X.; Zhao, H.Y.; Zhang, F.C.; Sun, Y.; Xiong, Z.Y.; Jiang, X.B. Dendritic cell-based vaccine for the treatment of malignant glioma: A systematic review. Cancer Invest. 2014, 32, 451–457, doi:10.3109/07357907.2014.958234.
- Eagles, M.E.; Nassiri, F.; Badhiwala, J.H.; Suppiah, S.; Almenawer, S.A.; Zadeh, G.; Aldape, K.D. Dendritic cell vac-cines for high-grade gliomas. Clin. Risk Manag. 2018, 14, 1299–1313, doi:10.2147/TCRM.S135865.
- Vatu, B.I.; Artene, S.A.; Staicu, A.G.; Turcu-Stiolica, A.; Folcuti, C.; Dragoi, A.; Cioc, C.; Baloi, S.C.; Tataranu, L.G.; Silosi, C.; et al. Assessment of efficacy of dendritic cell therapy and viral therapy in high grade glioma clinical trials. A meta-analytic review. J. Immunoass. Immunochem. 2019, 40, 70–80, doi:10.1080/15321819.2018.1551804.
- Lv, L.; Huang, J.; Xi, H.; Zhou, X. Efficacy and safety of dendritic cell vaccines for patients with glioblastoma: A me-ta-analysis of randomized controlled trials. Int. Immunopharmacol. 2020, 83, 106336, doi:10.1016/j.intimp.2020.106336.
- Burnet, N.G.; Jefferies, S.J.; Benson, R.J.; Hunt, D.P.; Treasure, F.P. Years of life lost (YLL) from cancer is an important measure of population burden--and should be considered when allocating research funds. Br. J. Cancer 2005, 92, 241–245.
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus ra-diotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the Eortc-Ncic trial. Lancet Oncol. 2009, 10, 459–466.
- Rouse, C.; Gittleman, H.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Years of potential life lost for brain and CNS tumors relative to other cancers in adults in the United States, 2010. Neuro Oncol. 2016, 18, 70–77, doi:10.1093/neuonc/nov249.
- Walker, M.D.; Strike, T.A.; Sheline, G.E. An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1725–1731, doi:10.1016/0360-3016(79)90553-4.
- Walker, M.D.; Alexander, E., Jr.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S., Jr.; Mealey, J., Jr.; Norrell, H.A.; Owens, G.; Ransohoff, J.; Wilson, C.B.; et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic glio-mas. A cooperative clinical trial. J. Neurosurg. 1978, 49, 333–343, doi:10.3171/jns.1978.49.3.0333.
- Gzell, C.; Back, M.; Wheeler, H.; Bailey, D.; Foote, M. Radiotherapy in Glioblastoma: The Past, the Present and the Future. Clin. Oncol. R Coll. Radiol. 2017, 29, 15–25, doi:10.1016/j.clon.2016.09.015.
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. New Engl. J. Med. 2005, 352, 997–1003.
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Me-sirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010, 17, 98–110, doi:10.1016/j.ccr.2009.12.020.
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glio-blastoma. Cancer Cell. 2012, 22, 425–437, doi:10.1016/j.ccr.2012.08.024.
- Dix, A.R.; Brooks, W.H.; Roszman, T.L.; Morford, L.A. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol. 1999, 100, 216–232.
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2020, doi:10.1007/s11060-020-03483-y.
- North, R.J. Gamma-irradiation facilitates the expression of adoptive immunity against established tumors by elim-inating suppressor T cells. Cancer Immunol. Immunother. 1984, 16, 175–181.
- Jordan, J.T.; Sun, W.; Hussain, S.F.; DeAngulo, G.; Prabhu, S.S.; Heimberger, A.B. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 2008, 57, 123–131.
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Cote, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multi-forme. Neuro Oncol. 2011, 13, 393–400, doi:10.1093/neuonc/noq204.
- Dutoit, V.; Philippin, G.; Widmer, V.; Marinari, E.; Vuilleumier, A.; Migliorini, D.; Schaller, K.; Dietrich, P.Y. Impact of Radiochemotherapy on Immune Cell Subtypes in High-Grade Glioma Patients. Front. Oncol. 2020, 10, 89, doi:10.3389/fonc.2020.00089.
- Antonopoulos, M.; Van Gool, S.W.; Dionysiou, D.; Graf, N.; Stamatakos, G. Immune phenotype correlates with sur-vival in patients with GBM treated with standard temozolomide-based therapy and immunotherapy. Anticancer Res. 2019, 39, 2043–2051.
- Sathornsumetee, S.; Rich, J.N. Designer therapies for glioblastoma multiforme. Ann. N. Y. Acad. Sci. 2008, 1142, 108–132.
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients with High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42, doi:10.1007/s11864-016-0418-0.
- dos Santos, M.A.; Pignon, J.P.; Blanchard, P.; Lefeuvre, D.; Levy, A.; Touat, M.; Louvel, G.; Dhermain, F.; Soria, J.C.; Deutsch, E.; et al. Systematic review and meta-analysis of phase I/II targeted therapy combined with radiotherapy in patients with glioblastoma multiforme: Quality of report, toxicity, and survival. J. Neurooncol. 2015, 123, 307–314, doi:10.1007/s11060-015-1802-5.
- Su, J.; Cai, M.; Li, W.; Hou, B.; He, H.; Ling, C.; Huang, T.; Liu, H.; Guo, Y. Molecularly Targeted Drugs Plus Radio-therapy and Temozolomide Treatment for Newly Diagnosed Glioblastoma: A Meta-Analysis and Systematic Re-view. Oncol. Res. 2016, 24, 117–128, doi:10.3727/096504016X14612603423511.
- Cihoric, N.; Tsikkinis, A.; Minniti, G.; Lagerwaard, F.J.; Herrlinger, U.; Mathier, E.; Soldatovic, I.; Jeremic, B.; Ghadjar, P.; Elicin, O.; et al. Current status and perspectives of interventional clinical trials for glioblastoma-analysis of Clin-icalTrials.gov. Radiat. Oncol. 2017, 12, 1, doi:10.1186/s13014-016-0740-5.
- Sim, H.W.; Morgan, E.R.; Mason, W.P. Contemporary management of high-grade gliomas. Cns Oncol. 2018, 7, 51–65, doi:10.2217/cns-2017-0026.
- Lavacchi, D.; Roviello, G.; D'Angelo, A. Tumor-Agnostic Treatment for Cancer: When How is Better than Where. Clin. Drug Investig. 2020, 40, 519–527, doi:10.1007/s40261-020-00915-5.
- Streptomycin treatment of pulmonary tuberculosis. Br. Med. J. 1948, 2, 769–782.
- Sorensen, H.T.; Lash, T.L.; Rothman, K.J. Beyond randomized controlled trials: A critical comparison of trials with nonrandomized studies. Hepatology 2006, 44, 1075–1082, doi:10.1002/hep.21404.
- Shikata, S.; Nakayama, T.; Noguchi, Y.; Taji, Y.; Yamagishi, H. Comparison of effects in randomized controlled trials with observational studies in digestive surgery. Ann. Surg. 2006, 244, 668–676, doi:10.1097/01.sla.0000225356.04304.bc.
- Vincent, J.L. We should abandon randomized controlled trials in the intensive care unit. Crit. Care Med. 2010, 38, S534–8, doi:10.1097/CCM.0b013e3181f208ac.
- Jones, D.S.; Podolsky, S.H. The history and fate of the gold standard. Lancet 2015, 385, 1502–1503, doi:10.1016/S0140-6736(15)60742-5.
- Trentino, K.; Farmer, S.; Gross, I.; Shander, A.; Isbister, J. Observational studies-should we simply ignore them in assessing transfusion outcomes? BMC Anesth. 2016, 16, 96, doi:10.1186/s12871-016-0264-4.
- Sharma, M.; Nazareth, I.; Petersen, I. Observational studies of treatment effectiveness: Worthwhile or worthless? Clin. Epidemiol. 2019, 11, 35–42, doi:10.2147/CLEP.S178723.
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, doi:10.1038/s41571-019-0177-5.
- Halabi, S.; Michiels, S. (Eds.) Textbook of Clinical trials in Oncology. A Statistical Perspective; CRC Press, Taylor & Fran-cis Group: New York, NY, USA, 2019.
- Stepanenko, A.A.; Chekhonin, V.P. Recent Advances in Oncolytic Virotherapy and Immunotherapy for Glio-blastoma: A Glimmer of Hope in the Search for an Effective Therapy? Cancers 2018, 10, doi:10.3390/cancers10120492.
- Walker, M.D.; Alexander, E., Jr.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S., Jr.; Mealey, J., Jr.; Norrell, H.A.; Owens, G.; Ransohoff, J.; Wilson, C.B.; et al. Evaluation of BCNU and/or radiotherapy in the treatment of ana-plastic gliomas. A cooperative clinical trial. J. Neurosurg. 1978, 49, 333–343, doi:10.3171/jns.1978.49.3.0333.
- Dix, A.R.; Brooks, W.H.; Roszman, T.L.; Morford, L.A. Immune defects observed in patients with primary ma-lignant brain tumors. J. Neuroimmunol. 1999, 100, 216–232.
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419, doi:10.3389/fonc.2018.00419.
- Suter, R.; Rodriguez-Blanco, J.; Ayad, N.G. Epigenetic pathways and plasticity in brain tumors. Neurobiol. Dis. 2020, 105060, doi:10.1016/j.nbd.2020.105060.
- Muller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br. J. Cancer 2020, 122, 295–305, doi:10.1038/s41416-019-0603-6.
- Fiegl, H.; Millinger, S.; Mueller-Holzner, E.; Marth, C.; Ensinger, C.; Berger, A.; Klocker, H.; Goebel, G.; Widschwendter, M. Circulating tumor-specific DNA: A marker for monitoring efficacy of adjuvant therapy in cancer patients. Cancer Res. 2005, 65, 1141–1145, doi:10.1158/0008-5472.CAN-04-2438.
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214, doi:10.1016/j.cell.2015.03.030.
- Macdonald, D.R.; Cascino, T.L.; Schold, S.C., Jr.; Cairncross, J.G. Response criteria for phase II studies of su-pratentorial malignant glioma. J. Clin. Oncol. 1990, 8, 1277–1280.
- Jaspan, T.; Morgan, P.S.; Warmuth-Metz, M.; Sanchez Aliaga, E.; Warren, D.; Calmon, R.; Grill, J.; Hargrave, D.; Garcia, J.; Zahlmann, G. Response Assessment in Pediatric Neuro-Oncology: Implementation and Expansion of the RANO Criteria in a Randomized Phase II Trial of Pediatric Patients with Newly Diagnosed High-Grade Gliomas. Am. J. Neuroradiol. 2016, 37, 1581–1587, doi:10.3174/ajnr.A4782.
- Okada, H.; Weller, M.; Huang, R.; Finocchiaro, G.; Gilbert, M.R.; Wick, W.; Ellingson, B.M.; Hashimoto, N.; Pol-lack, I.F.; Brandes, A.A.; et al. Immunotherapy response assessment in neuro-oncology: A report of the RANO working group. Lancet Oncol. 2015, 16, e534–e542, doi:10.1016/S1470-2045(15)00088-1.
- Aquino, D.; Gioppo, A.; Finocchiaro, G.; Bruzzone, M.G.; Cuccarini, V. MRI in Glioma Immunotherapy: Evi-dence, Pitfalls, and Perspectives. J. Immunol. Res. 2017, 2017, 5813951, doi:10.1155/2017/5813951.
- Sharma, P.; Debinski, W. Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 2018, 19, doi:10.3390/ijms19113326.
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharm. 2017, 8, 242, doi:10.3389/fphar.2017.00242.
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 87, doi:10.1186/s13046-019-1085-3.
- Ameratunga, M.; Coleman, N.; Welsh, L.; Saran, F.; Lopez, J. CNS cancer immunity cycle and strategies to tar-get this for glioblastoma. Oncotarget 2018, 9, 22802–22816, doi:10.18632/oncotarget.24896.
- Zhu, C.; Zou, C.; Guan, G.; Guo, Q.; Yan, Z.; Liu, T.; Shen, S.; Xu, X.; Chen, C.; Lin, Z.; et al. Development and validation of an interferon signature predicting prognosis and treatment response for glioblastoma. Oncoim-munology 2019, 8, e1621677, doi:10.1080/2162402X.2019.1621677.
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite inte-grated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroim-munol. 2013, 264, 71–83, doi:10.1016/j.jneuroim.2013.08.013.
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting from Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211, doi:10.1200/JCO.2016.66.6552.
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, doi:10.3390/cells9020263.
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. p53 increases MHC class I expression by upregulating the endoplasmic re-ticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359, doi:10.1038/ncomms3359.
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. Cancer Immunology. The cancer immunogram. Science 2016, 352, 658–660, doi:10.1126/science.aaf2834.
- Macdonald, D.R.; Cascino, T.L.; Schold, S.C., Jr.; Cairncross, J.G. Response criteria for phase II studies of supratentorial malignant glioma. J. Clin. Oncol. 1990, 8, 1277–1280.
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Micro-environment and Host State-Implications for Immunotherapy. Clin. Cancer Res. 2019, doi:10.1158/1078-0432.CCR-18-1627.
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689, doi:10.3390/ijms21030689.
- Zhang, B.; Shen, R.; Cheng, S.; Feng, L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 2019, 8, 2897–2907, doi:10.1002/cam4.2192.
- Hallaert, G.; Pinson, H.; Van den Broecke, C.; Vanhauwaert, D.; Van Roost, D.; Boterberg, T.; Kalala, J.P. Subventricular zone contacting glioblastoma: Tumor size,
- molecular biological factors and patient survival. Acta Oncol. 2020, 1–6, doi:10.1080/0284186X.2020.1794032.
- Berendsen, S.; van Bodegraven, E.; Seute, T.; Spliet, W.G.M.; Geurts, M.; Hendrikse, J.; Schoysman, L.; Huiszoon, W.B.; Varkila, M.; Rouss, S.; et al. Adverse prognosis of glioblastoma contacting the subventricular zone: Biological correlates. PLoS ONE 2019, 14, e0222717, doi:10.1371/journal.pone.0222717.
- Wee, C.W.; Kim, I.H.; Park, C.K.; Kim, J.W.; Dho, Y.S.; Ohka, F.; Aoki, K.; Motomura, K.; Natsume, A.; Kim, N.; et al. Validation of a novel molecular RPA classification in glioblastoma (GBM-molRPA) treated with chemoradiation: A multi-institutional collaborative study. Radiother. Oncol. 2018, 129, 347–351, doi:10.1016/j.radonc.2018.09.001.
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072, doi:10.1158/1078-0432.CCR-15-0428.
- O'Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, doi:10.1126/scitranslmed.aaa0984.
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene. 2018, doi:10.1038/s41417-018-0062-x.
- Rammensee, H.G.; Singh-Jasuja, H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev. Vaccines 2013, 12, 1211–1217, doi:10.1586/14760584.2013.836911.
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, doi:10.1038/nature23003.
- Johanns, T.M.; Miller, C.A.; Liu, C.J.; Perrin, R.J.; Bender, D.; Kobayashi, D.K.; Campian, J.L.; Chicoine, M.R.; Dacey, R.G.; Huang, J.; et al. Detection of neoantigen-specific T cells following a personalized vaccine in a patient with glioblastoma. Oncoimmunology 2019, 8, e1561106, doi:10.1080/2162402X.2018.1561106.
- De Vleeschouwer, S.; Arredouani, M.; Ade, M.; Cadot, P.; Vermassen, E.; Ceuppens, J.L.; Van Gool, S.W. Uptake and presentation of malignant glioma tumor cell lysates by monocyte-derived dendritic cells. Cancer Immunol. Immunother. 2005, 54, 372–382.
- De Vleeschouwer, S.; Spencer, L.I.; Ceuppens, J.L.; Van Gool, S.W. Persistent IL-10 production is required for glioma growth suppressive activity by Th1-directed effector cells after stimulation with tumor lysate-loaded dendritic cells. J. Neurooncol. 2007, 84, 131–140.
- Maes, W.; Deroose, C.; Reumers, V.; Krylyshkina, O.; Gijsbers, R.; Ceuppens, J.; Baekelandt, V.; Debyser, Z.; Van Gool, S. In vivo bioluminescence imaging in an experimental mouse model for dendritic cell based immunotherapy against malignant glioma. J. Neuro Oncol. 2009, 91, 127–139.
- Maes, W.; Galicia Rosas, G.; Verbinnen, B.; Boon, L.; De Vleeschouwer, S.; Ceuppens, J.L.; Van Gool, S.W. DC vaccination with anti-CD25 treatment leads to long-term immunity against experimental glioma. Neuro Oncol. 2009, 11, 529–542.
- Ardon, H.; Verbinnen, B.; Maes, W.; Beez, T.; Van Gool, S.; De Vleeschouwer, S. Technical advancement in regulatory T cell isolation and characterization using CD127 expression in patients with malignant glioma treated with autologous dendritic cell vaccination. J. Immunol. Methods 2010, 352, 169–173.
- Maes, W.; Van Gool, S.W. Experimental immunotherapy for malignant glioma: Lessons from two decades of research in the GL261 model. Cancer Immunol. Immunother. 2011, 60, 153–160, doi:10.1007/s00262-010-0946-6.
- Vandenberk, L.; Van Gool, S.W. Treg infiltration in glioma: A hurdle for antiglioma immunotherapy. Immunotherapy 2012, 4, 675–678, doi:10.2217/imt.12.64.
- Koks, C.A.E.; Garg, A.D.; Ehrhardt, M.; Riva, M.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2014, 136, e313–e325.
- Koks, C.A.; De Vleeschouwer, S.; Graf, N.; Van Gool, S.W. Immune Suppression during Oncolytic Virotherapy for High-Grade Glioma; Yes or No? J. Cancer 2015, 6, 203–217, doi:10.7150/jca.10640.
- Vandenberk, L.; Belmans, J.; Van Woensel, M.; Riva, M.; Van Gool, S.W. Exploiting the Immunogenic Potential of Cancer Cells for Improved Dendritic Cell Vaccines. Front. Immunol. 2015, 6, 663, doi:10.3389/fimmu.2015.00663.
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra327, doi:10.1126/scitranslmed.aae0105.
- Vandenberk, L.; Garg, A.D.; Verschuere, T.; Koks, C.; Belmans, J.; Beullens, M.; Agostinis, P.; De Vleeschouwer, S.; Van Gool, S.W. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology 2016, 5, e1083669, doi:10.1080/2162402X.2015.1083669.
- Belmans, J.; Van Woensel, M.; Creyns, B.; Dejaegher, J.; Bullens, D.M.; Van Gool, S.W. Immunotherapy with subcutaneous immunogenic autologous tumor lysate increases murine glioblastoma survival. Sci. Rep. 2017, 7, 13902, doi:10.1038/s41598-017-12584-0.
- Liau, L.M.; Black, K.L.; Martin, N.A.; Sykes, S.N.; Bronstein, J.M.; Jouben-Steele, L.; Mischel, P.S.; Belldegrun, A.; Cloughesy, T.F. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Case Report. Neurosurg. Focus 2000, 9, e8.
- Yu, J.S.; Wheeler, C.J.; Zeltzer, P.M.; Ying, H.; Finger, D.N.; Lee, P.K.; Yong, W.H.; Incardona, F.; Thompson, R.C.; Riedinger, M.S.; et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001, 61, 842–847.
- Kikuchi, T.; Akasaki, Y.; Irie, M.; Homma, S.; Abe, T.; Ohno, T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol. Immunother. 2001, 50, 337–344.
- Wheeler, C.J.; Black, K.L.; Liu, G.; Ying, H.; Yu, J.S.; Zhang, W.; Lee, P.K. Thymic CD8(+) T cell production strongly influences tumor antigen recognition and age-dependent glioma mortality. J. Immunol. 2003, 171, 4927–4933.
- Yamanaka, R.; Abe, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Kobayashi, T.; Narita, M.; Takahashi, M.; Tanaka, R. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: Results of a clinical phase I/II trial. Br. J. Cancer 2003, 89, 1172–1179.
- Caruso, D.A.; Orme, L.M.; Neale, A.M.; Radcliff, F.J.; Amor, G.M.; Maixner, W.; Downie, P.; Hassall, T.E.; Tang, M.L.; Ashley, D.M. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro. Oncol. 2004, 6, 236–246, doi:10.1215/S1152851703000668.
- De Vleeschouwer, S.; Van Calenbergh, F.; Demaerel, P.; Flamen, P.; Rutkowski, S.; Kaempgen, E.; Wolff, J.E.A.; Plets, C.; Sciot, R.; Van Gool, S.W. Transient local response and persistent tumor control of recurrent malignant glioma treated with combination therapy including dendritic cell therapy. J. Neurosurg. 2004, 100, 492–497.
- Kikuchi, T.; Akasaki, Y.; Abe, T.; Fukuda, T.; Saotome, H.; Ryan, J.L.; Kufe, D.W.; Ohno, T. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J. Immunother. 2004, 27, 452–459.
- Rutkowski, S.; De Vleeschouwer, S.; Kaempgen, E.; Wolff, J.E.A.; Kuhl, J.; Demaerel, P.; Warmuth-Metz, M.; Flamen, P.; Van Calenbergh, F.; Plets, C.; et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br. J. Cancer 2004, 91, 1656–1662.
- Wheeler, C.J.; Das, A.; Liu, G.; Yu, J.S.; Black, K.L. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin. Cancer Res. 2004, 10, 5316–5326.
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004, 64, 4973–4979.
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525.
- Yamanaka, R.; Honma, J.; Tsuchiya, N.; Yajima, N.; Kobayashi, T.; Tanaka, R. Tumor lysate and IL-18 loaded dendritic cells elicits Th1 response, tumor-specific CD8+ cytotoxic T cells in patients with malignant glioma. J. Neurooncol. 2005, 72, 107–113.
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M.; et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: Results of a clinical phase I/II trial. Clin. Cancer Res. 2005, 11, 4160–4167.
- Khan, J.A.; Yaqin, S. Dendritic cell therapy with improved outcome in glioma multiforme—A case report. J. Zhejiang Univ. Sci. B 2006, 7, 114–117.
- Okada, H.; Lieberman, F.S.; Walter, K.A.; Lunsford, L.D.; Kondziolka, D.S.; Bejjani, G.K.; Hamilton, R.L.; Torres-Trejo, A.; Kalinski, P.; Cai, Q.; et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J. Transl. Med. 2007, 5, 67.
- De Vleeschouwer, S.; Fieuws, S.; Rutkowski, S.; Van Calenbergh, F.; Van Loon, J.; Goffin, J.; Sciot, R.; Wilms, G.; Demaerel, P.; Warmuth-Metz, M.; et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 3098–3104.
- Prins, R.M.; Cloughesy, T.F.; Liau, L.M. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. New Engl. J. Med. 2008, 359, 539–541, doi:10.1056/NEJMc0804818.
- Walker, D.G.; Laherty, R.; Tomlinson, F.H.; Chuah, T.; Schmidt, C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: Potential interaction with adjuvant chemotherapy. J. Clin. Neurosci. 2008, 15, 114–121.
- Wheeler, C.J.; Black, K.L.; Liu, G.; Mazer, M.; Zhang, X.X.; Pepkowitz, S.; Goldfinger, D.; Ng, H.; Irvin, D.; Yu, J.S. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008, 68, 5955–5964.
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E.; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer 2009, 8, 2773–2779, doi:10.1158/1535-7163.MCT-09-0124.
- Ardon, H.; Van Gool, S.; Lopes, I.S.; Maes, W.; Sciot, R.; Wilms, G.; Demaerel, P.; Bijttebier, P.; Claes, L.; Goffin, J.; et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: A pilot study. J. Neurooncol. 2010, 99, 261–272.
- Ardon, H.; De Vleeschouwer, S.; Van Calenbergh, F.; Claes, L.; Kramm, C.M.; Rutkowski, S.; Wolff, J.E.; Van Gool, S.W. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr. Blood Cancer 2010, 54, 519–525.
- Chang, C.N.; Huang, Y.C.; Yang, D.M.; Kikuta, K.; Wei, K.J.; Kubota, T.; Yang, W.K. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J. Clin. Neurosci. 2011, 18, 1048–1054, doi:10.1016/j.jocn.2010.11.034.
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 2011, 34, 382–389, doi:10.1097/CJI.0b013e318215e300.
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011, 29, 330–336, doi:10.1200/JCO.2010.30.7744.
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615, doi:10.1158/1078-0432.CCR-10-2563.
- Akiyama, Y.; Oshita, C.; Kume, A.; Iizuka, A.; Miyata, H.; Komiyama, M.; Ashizawa, T.; Yagoto, M.; Abe, Y.; Mitsuya, K.; et al. alpha-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: A phase I clinical trial. BMC Cancer 2012, 12, 623, doi:10.1186/1471-2407-12-623 [doi].
- Ardon, H.; Van Gool, S.W.; Verschuere, T.; Maes, W.; Fieuws, S.; Sciot, R.; Wilms, G.; Demaerel, P.; Goffin, J.; Van Calenbergh, F.; et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: Results of the HGG-2006 phase I/II trial. Cancer Immunol. Immunother. 2012, 61, 2033–2044, doi:10.1007/s00262-012-1261-1.
- Cho, D.Y.; Yang, W.K.; Lee, H.C.; Hsu, D.M.; Lin, H.L.; Lin, S.Z.; Chen, C.C.; Harn, H.J.; Liu, C.L.; Lee, W.Y.; et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: A phase II clinical trial. World Neurosurg. 2012, 77, 736–744, doi:10.1016/j.wneu.2011.08.020.
- de Vleeschouwer, S.; Ardon, H.; van Calenbergh, F.; Sciot, R.; Wilms, G.; van Loon, J.; Goffin, J.; van Gool, S. Stratification according to Hgg-Immuno Rpa model predicts outcome in a large group of patients with relapsed malignant glioma treated by adjuvant postoperative dendritic cell vaccination. Cancer Immunol. Immunother. 2012, 61, 2105–2112, doi:10.1007/s00262-012-1271-z.
- Elens, I.; De Vleeschouwer, S.; Pauwels, F.; Van Gool, S.W. Resection and immunotherapy for recurrent grade III glioma. Isrn Immunol. 2012, 2012, 1–9.
- Fong, B.; Jin, R.; Wang, X.; Safaee, M.; Lisiero, D.N.; Yang, I.; Li, G.; Liau, L.M.; Prins, R.M. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS ONE 2012, 7, e32614, doi:10.1371/journal.pone.0032614.
- Iwami, K.; Shimato, S.; Ohno, M.; Okada, H.; Nakahara, N.; Sato, Y.; Yoshida, J.; Suzuki, S.; Nishikawa, H.; Shiku, H.; et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy 2012, doi:10.3109/14653249.2012.666633.
- Jie, X.; Hua, L.; Jiang, W.; Feng, F.; Feng, G.; Hua, Z. Clinical application of a dendritic cell vaccine raised against heat-shocked glioblastoma. Cell Biochem. Biophys. 2012, 62, 91–99, doi:10.1007/s12013-011-9265-6.
- Qin, K.; Tian, G.; Li, P.; Chen, Q.; Zhang, R.; Ke, Y.Q.; Xiao, Z.C.; Jiang, X.D. Anti-glioma response of autologous T cells stimulated by autologous dendritic cells electrofused with CD133(+) or CD133(-) glioma cells. J. Neuroimmunol. 2012, 242, 9–15, doi:10.1016/j.jneuroim.2011.11.001.
- Sampson, J.H.; Schmittling, R.J.; Archer, G.E.; Congdon, K.L.; Nair, S.K.; Reap, E.A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E.; et al. A Pilot Study of IL-2Ralpha Blockade during Lymphopenia Depletes Regulatory T-cells and Correlates with Enhanced Immunity in Patients with Glioblastoma. PLoS ONE 2012, 7, e31046, doi:10.1371/journal.pone.0031046.
- Valle, R.D.; de Cerio, A.L.; Inoges, S.; Tejada, S.; Pastor, F.; Villanueva, H.; Gallego, J.; Espinos, J.; Aristu, J.; Idoate, M.A.; et al. Dendritic cell vaccination in glioblastoma after fluorescence-guided resection. World J. Clin. Oncol. 2012, 3, 142–149, doi:10.5306/wjco.v3.i11.142.
- Lasky, J.L., III; Panosyan, E.H.; Plant, A.; Davidson, T.; Yong, W.H.; Prins, R.M.; Liau, L.M.; Moore, T.B. Autolo-gous Tumor Lysate-pulsed Dendritic Cell Immunotherapy for Pediatric Patients with Newly Diagnosed or Re-current High-grade Gliomas. Anticancer Res. 2013, 33, 2047–2056, doi:33/5/2047.
- Pellegatta, S.; Eoli, M.; Frigerio, S.; Antozzi, C.; Bruzzone, M.G.; Cantini, G.; Nava, S.; Anghileri, E.; Cuppini, L.; Cuccarini, V.; et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoim-munology 2013, 2, e23401, doi:10.4161/onci.23401.
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135, doi:10.1007/s00262-012-1319-0.
- Prins, R.M.; Wang, X.; Soto, H.; Young, E.; Lisiero, D.N.; Fong, B.; Everson, R.; Yong, W.H.; Lai, A.; Li, G.; et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J. Immunother. 2013, 36, 152–157, doi:10.1097/CJI.0b013e3182811ae4.
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tonnesen, P.; Suso, E.M.; Saeboe-Larssen, S.; Sand-berg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, doi:10.1007/s00262-013-1453-3.
- Eyrich, M.; Schreiber, S.C.; Rachor, J.; Krauss, J.; Pauwels, F.; Hain, J.; Wolfl, M.; Lutz, M.B.; De, V.S.; Schlegel, P.G.; et al. Development and validation of a fully GMP-compliant production process of autologous, tu-mor-lysate-pulsed dendritic cells. Cytotherapy 2014, 16, 946–964, doi:10.1016/j.jcyt.2014.02.017.
- Van Gool, S.W.; Makalowski, J.; Feyen, O.; Prix, L.; Schirrmacher, V.; Stuecker, W. The induction of immuno-genic cell death (ICD) during maintenance chemotherapy and subsequent multimodal immunotherapy for glioblastoma (GBM). Austin Oncol. Case Rep. 2018, 3, 1010.
- Ishikawa, E.; Muragaki, Y.; Yamamoto, T.; Maruyama, T.; Tsuboi, K.; Ikuta, S.; Hashimoto, K.; Uemae, Y.; Ishi-hara, T.; Matsuda, M.; et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous for-malin-fixed tumor vaccine for newly diagnosed glioblastoma. J. Neurosurg. 2014, 121, 543–553, doi:10.3171/2014.5.JNS132392.
- Hunn, M.K.; Bauer, E.; Wood, C.E.; Gasser, O.; Dzhelali, M.; Ancelet, L.R.; Mester, B.; Sharples, K.J.; Findlay, M.P.; Hamilton, D.A.; et al. Dendritic cell vaccination combined with temozolomide retreatment: Results of a phase I trial in patients with recurrent glioblastoma multiforme. J. Neurooncol. 2015, 121, 319–329, doi:10.1007/s11060-014-1635-7.
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glio-blastoma patients. Nature 2015, 519, 366–369, doi:10.1038/nature14320.
- Muller, K.; Henke, G.; Pietschmann, S.; van Gool, S.; De Vleeschouwer, S.; von Bueren, A.O.; Compter, I.; Friedrich, C.; Matuschek, C.; Klautke, G.; et al. Re-irradiation or re-operation followed by dendritic cell vac-cination? Comparison of two different salvage strategies for relapsed high-grade gliomas by means of a new prognostic model. J. Neurooncol. 2015, 124, 325–332, doi:10.1007/s11060-015-1844-8.
- Sakai, K.; Shimodaira, S.; Maejima, S.; Udagawa, N.; Sano, K.; Higuchi, Y.; Koya, T.; Ochiai, T.; Koide, M.; Uehara, S.; et al. Dendritic cell-based immunotherapy targeting Wilms' tumor 1 in patients with recurrent ma-lignant glioma. J. Neurosurg. 2015, 123, 989–997, doi:10.3171/2015.1.JNS141554.
- Van Gool, S.W. Brain tumor immunotherapy: What have we learned so far? Front. Oncol. 2015, 5, 98, doi:10.3389/fonc.2015.00098.
- Akasaki, Y.; Kikuchi, T.; Homma, S.; Koido, S.; Ohkusa, T.; Tasaki, T.; Hayashi, K.; Komita, H.; Watanabe, N.; Suzuki, Y.; et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fu-sions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol. Immunother. 2016, 65, 1499–1509, doi:10.1007/s00262-016-1905-7.
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Normolle, D.P.; Connelly, A.K.; Dibridge, S.; Mason, G.; Whiteside, T.L.; et al. Antigen-specific immunoreactivity and clinical outcome follow-ing vaccination with glioma-associated antigen peptides in children with recurrent high-grade gliomas: Results of a pilot study. J. Neurooncol. 2016, 130, 517–527, doi:10.1007/s11060-016-2245-3.
- Inoges, S.; Tejada, S.; de Cerio, A.L.; Gallego Perez-Larraya, J.; Espinos, J.; Idoate, M.A.; Dominguez, P.D.; de Eulate, R.G.; Aristu, J.; Bendandi, M.; et al. A phase II trial of autologous dendritic cell vaccination and radi-ochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J. Transl. Med. 2017, 15, 104, doi:10.1186/s12967-017-1202-z.
- Sakai, K.; Shimodaira, S.; Maejima, S.; Sano, K.; Higuchi, Y.; Koya, T.; Sugiyama, H.; Hongo, K. Clinical effect and immunological response in patients with advanced malignant glioma treated with WT1-pulsed dendritic cell-based immunotherapy: A report of two cases. Interdiscip. Neurosurg. Adv. Tech. Case Manag. 2017, 9, 24–29.
- Benitez-Ribas, D.; Cabezon, R.; Florez-Grau, G.; Molero, M.C.; Puerta, P.; Guillen, A.; Paco, S.; Carcaboso, A.M.; Santa-Maria Lopez, V.; Cruz, O.; et al. Immune Response Generated with the Administration of Autologous Dendritic Cells Pulsed with an Allogenic Tumoral Cell-Lines Lysate in Patients with Newly Diagnosed Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 127, doi:10.3389/fonc.2018.00127.
- Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.F.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Can-cers 2018, 10, doi:10.3390/cancers10100372.
- Jan, C.I.; Tsai, W.C.; Harn, H.J.; Shyu, W.C.; Liu, M.C.; Lu, H.M.; Chiu, S.C.; Cho, D.Y. Predictors of Response to Autologous Dendritic Cell Therapy in Glioblastoma Multiforme. Front. Immunol. 2018, 9, 727, doi:10.3389/fimmu.2018.00727.
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: Immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovaria-bles. Acta Neuropathol. Commun. 2018, 6, 135, doi:10.1186/s40478-018-0621-2.
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D'Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142, doi:10.1186/s12967-018-1507-6.


