 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nozie Dominic Aghaizu | + 2796 word(s) | 2796 | 2021-01-05 09:43:03 | | | |
| 2 | Peter Tang | -98 word(s) | 2698 | 2021-01-23 09:46:42 | | |
Video Upload Options
The Wnt signalling system is essential for both the developing and adult central nervous system. It regulates numerous cellular functions ranging from neurogenesis to blood brain barrier biology. Dysregulated Wnt signalling can thus have significant consequences for normal brain function, which is becoming increasingly clear in Alzheimer's disease (AD), an age-related neurodegenerative disorder that is the most prevalent form of dementia. AD exhibits a range of pathophysiological manifestations including aberrant amyloid precursor protein processing, tau pathology, synapse loss, neuroinflammation and blood brain barrier breakdown, which have been associated to a greater or lesser degree with abnormal Wnt signalling.
1. Introduction
The term dementia encompasses a group of devastating disorders with characteristic declines in cognition, function and behaviour, which eventually severely impair patients' ability to perform instrumental and/or basic activities [1][2]. Beyond the patient, this profoundly impacts caregivers, families and society as a whole, which is facing ever increasing health care costs associated with dementia [3]. Alzheimer's disease (AD) is by far the most common form of dementia accounting for 60–80% of cases [3], with an estimated 40 million diagnosed patients worldwide [4]. Current therapeutic options are restricted to symptomatic treatments such as cholinesterase inhibitors or the glutamate receptor antagonist memantine, which have a limited effect on memory and cognition [5]. As disease-modifying therapies, which target the disease process, are not yet available [6][7], AD dementia inevitably results in death within 5–12 years of symptom onset [8].
While the need for disease-modifying therapies that prevent disease onset or slow disease progression is clear, the lack of such treatments reflects the complicated, multifactorial pathobiology of AD. Patients often present with a range of disease manifestations, but what uniquely defines AD is the presence of protein aggregates in the form of extracellular deposits of β-amyloid (Aβ) as diffuse and neuritic plaques, as well as intracellular neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau [9] observed in post-mortem tissue. Yet, as of writing, therapeutic approaches targeting these two disease hallmarks have not proven to be effective in clinical trials [6][7] (although approaches targeting tau are at a relatively early stage of evaluation in patients [10]). This is likely to be for a number of reasons, beyond the scope of this review. However, it does raise the possibility that an effective therapeutic strategy will require the targeting of several pathological manifestations via different biological pathways, and that this may change over the time course of the disease.
The Wnt signalling system plays a crucial role in many cellular processes such as cell differentiation, migration, and tissue homeostasis. In the central nervous system (CNS), Wnt signalling regulates developmental programmes and, as is increasingly recognised, it modulates a number of aspects of the mature brain such as synapse number and function, the integrity and function of the blood brain barrier (BBB), as well as the biology of microglia (the resident immune cells of the CNS). These key facets of the mature brain are significantly impacted in AD, raising the possibility that dysregulation of Wnt signalling may play an important role in a number of different aspects of this complex disease [11][12].
2. Wnt Signalling Pathways—An Overview
Wnt ligands are lipid-modified [13], secreted glycoproteins, which, upon binding to cell surface receptors, trigger intracellular signalling pathways that regulate various biological processes such as the cell cycle, cell migration and establishment of cell polarity [14][15]. The genomes of most mammals harbour 19 Wnt genes that can be grouped into 12 conserved subfamilies. These ligands are recognised by a heterodimeric receptor complex on the cell surface comprising Frizzled (Fz) as well as single-pass transmembrane co-receptors LRP5, LRP6, Ror1, Ror2, or Ryk proteins (Figure 1) [16][17][18][19]. The mammalian genome harbours 10 Fz genes, which encode 7-transmembrane (7TM) receptors that exhibit an N-terminal, extracellular, large cysteine-rich domain (CRD) used for Wnt binding [20][21][22]. However, ligand-receptor interactions are promiscuous as evidenced by the fact that there are multiple, non-mutually exclusive ligand receptor combinations [23][24].
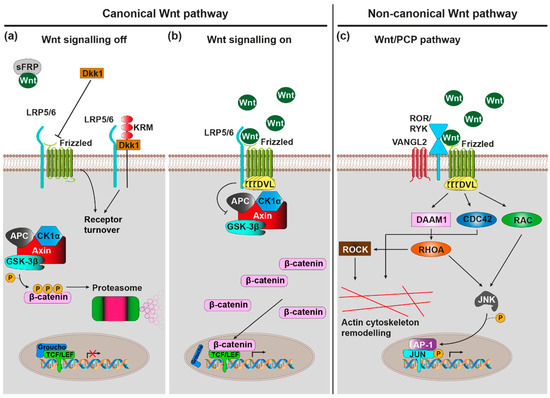
Figure 1. Wnt signalling pathways. Note that emphasis is placed on pathways and components that are of greater relevance in the context of AD. (a) Canonical Wnt signalling pathway, off: In absence of Wnt ligand binding to Frizzled (Fz)-LRP5/6 heterodimeric cell surface receptors, the Axin/APC/CK1α/GSK-3β destruction complex phosphorylates the transcription factor β-catenin, marking it for proteasomal degradation. Endogenous canonical Wnt antagonists (including sFRP and Dkk1) further favour the ‘off' state. (b) Canonical Wnt signalling pathway, on: Wnt ligand binding to Fz-LRP5/6 recruits the scaffolding protein DVL, which in turn inhibits GSK-3β activity of the destruction complex. β-catenin subsequently accumulates in cytoplasm, allowing it to translocate to the nucleus where it transactivates the TCF/LEF-mediated expression of canonical Wnt target genes. (c) Wnt/planar cell polarity (PCP) pathway: Wnt ligand binding to Fz-ROR/RYK co-receptors recruits DVL, which in turn activates the small GTPases RHOA, RAC, and CDC42. RHOA and RAC conjointly activate JNK, which stimulates Jun transcription factor dependent gene expression via phosphorylation. Furthermore, RHOA, via ROCK, and CDC42 stimulate actin cytoskeleton remodelling.
Wnt ligand binding by the receptor complex induces conformational changes of the receptor complex and phosphorylation of target proteins as intracellular signalling pathways are initiated. These signalling pathways can broadly be classified into the canonical Wnt/β-catenin and non-canonical β-catenin-independent signalling cascades. The activation of specific pathways depends on, among various other factors, the exact identity of involved ligand and receptor isoforms, the expression of which is under tight spatiotemporal control [23].
The canonical Wnt/β-catenin signalling cascade is initiated upon binding of Wnt to the Fz-LRP receptor complex (Figure 1a,b) [25]. This subsequently leads to the inactivation of a multiprotein complex consisting of CK1α, GSK-3β, Axin, and APC, which usually phosphorylates β-catenin, thus marking it for proteasomal degradation [26]. Accumulating β-catenin can now translocate to the nucleus where it associates with transcription factors from the TCF/Lef family [27][28] to activate transcription of Wnt/β-catenin target genes with known roles in proliferation, fate specification and differentiation in development as well as adult tissue homeostasis [29].
Initiation of the non-canonical Wnt/PCP (planar cell polarity) cascade requires Wnt binding to a Fz-Ror/Ryk receptor complex (Figure 1c) [30][31][32]. Intracellularly, this causes the activation of small GTPases RhoA, Rac, and Cdc42 [33][34], which subsequently activate the downstream kinases JNK and ROCK [35][36], which in turn regulate actin and microtubule cytoskeletons. As such, the Wnt/PCP cascade plays a vital role in the control of cell/tissue polarity and cell migration [37].
The Wnt/Ca2+ cascade requires binding of Wnt ligand to Fz receptor, which intracellularly triggers G-protein coupled signalling [38]. This in turn activates phospholipase C (PLC) [39], which stimulates the release of Ca2+ from intracellular stores via the signalling molecule inositol triphosphate (IP3) [40]. The mobilised Ca2+ then stimulates the Ca2+-sensitive protein kinases protein kinase C (PKC) [41] and Ca2+/Calmodulin-dependent protein kinase II (CaMKII) [42], as well as the Ca2+ sensitive transcription factor NF-AT. Through these effector proteins the Wnt/Ca2+ cascade regulates many processes, ranging from developmental cell fate determination, cell/tissue migration, cell differentiation, and inflammatory response mediation [43].
Wnt signalling can furthermore be modulated by a number of endogenous agonists and antagonists [44], which are important for the fine-tuning of Wnt signalling-regulated processes. There are seven secreted antagonist families (the Dickkopf proteins (Dkks), secreted Frizzled-related proteins (sFRPs), Wnt inhibitory factor 1 (WIF-1), Wise/SOST, Cerberus, insulin-like growth factor binding protein 4 (IGFBP-4), Notum and four transmembrane Wnt antagonist families (Shisa, Wnt-activated inhibitory factor 1 (Waif1/5T4), adenomatosis polyposis coli down-regulated 1 (APCDD1), and Tiki1). They exert their function either by sequestering/inactivating secreted Wnt (e.g., WIF1, Cerberus, sFRP, Notum) or by blocking/sequestering elements of receptor complexes (e.g., Dkk1, Wise/SOST, IGFBP-4). For example, Dkk1 sequesters LRP6, thus preventing its heterodimerisation with Fz8 to block canonical Wnt/β-catenin signalling [45]. When the Dkk1 co-receptor Kremen 2 (Krm2) is present, this additionally leads to the endocytosis of LRP5/6-Krm2-Dkk1 complexes [46]. The related Dkk2, however, can act both as an activator and as an inhibitor of the Wnt/β-catenin cascade [47], in the presence Krm2, Dkk2 functions as an LRP6 antagonist, while in its absence it functions as an activator.
Conversely, there are two families of secreted proteins that act purely as Wnt agonists: R-spondins (Rspo) and Norrin. R-spondins stimulate canonical Wnt/β-catenin signalling by promoting the internalisation of the transmembrane E3 ubiquitin ligase ZNRF3 that usually marks Fz and LRP6 for degradation [48]; this is mediated by the Rspo receptors Lgr4, Lgr5, and Lgr6 [49]. R-spondins can also stimulate Wnt/PCP signalling via a mechanism that requires Wnt5a-Fz7 signalling, Rspo3 binding to the four transmembrane proteoglycan syndecan 4, and syndecan 4-dependent endocytosis of the entire Wnt5a-Fz7-Rspo3-syndecan 4 complex [50]; in contrast to the action of Dkk1/Krm2 on canonical Wnt/β-catenin signalling through Fz8/LRP6, this internalisation was shown to be crucial for Wnt/PCP signal transduction. Finally, Norrin, although structurally unrelated to Wnts, binds Fz4/LRP5 to activate Wnt/β-catenin signalling [51].
The Wnt cascades thus constitute a highly complex signalling network with a range of different functions and roles in development, mature homeostasis, ageing, as well as disease. The remainder of this review will provide more detailed insight into Wnt signalling in the brain with a focus on the deregulation of Wnt signalling in AD.
3. Wnt Signalling in the Brain
Wnt signalling plays an important role in various aspects of the brain, ranging from brain development to normal brain function. Indeed, the gene expression of various Wnt and Fz receptor isoforms is subject to tight spatio-temporal control, as has been reviewed in [52]. Unsurprisingly, altered Wnt signalling strength can have detrimental effects on the brain, as is for instance observed during ageing where reduced Wnt signalling is evident.
4. Wnt Signalling in AD
Given the connection between age-related cognitive decline and decreased Wnt signalling strength, it is perhaps unsurprising that Wnt signalling pathways are also suppressed in AD given the strong connection between AD and its biggest risk factor–age [53][54]. Indeed, the majority of AD cases are detected at an advanced age (sporadic late onset AD or LOAD) supporting the fact the AD is predominantly an age-related disorder of the brain. However, mutations in certain genes result in early onset heritable/familial AD or FAD). While FAD is associated with mutations in amyloid precursor protein (APP) or APP processing genes (PSEN1, PSEN2, encoding components of the γ-secretase complex), numerous additional risk genes (including APOE, TREM2, and UNC5C) as well as environmental factors confer susceptibility to LOAD. Genetic studies have been invaluable in shedding light on the multiple pathological mechanisms observed in AD, as will be discussed below. In several cases where gene variants have been linked to AD, these genes also show connections to Wnt signalling pathways.
Among the first pieces of evidence indicating that abnormal Wnt signalling could be involved in AD came from a study that discovered a polymorphism in the GSK3β promoter, which increased its activity and conferred increased susceptibility to LOAD [55] (given the known biological role of GSK-3β, increased activity reduces canonical Wnt signalling by promoting β-catenin degradation). Further supporting the link between reduced canonical signalling and AD, a single nucleotide polymorphism (Ile-1062 → Val) and a novel splice variant within the canonical Fz co-receptor encoding gene LRP6 were also associated with LOAD; functionally, in HEK293T cells, both reduced canonical Wnt signalling [56][57]. In addition, LRP6 mRNA and protein levels were significantly decreased in human AD brains compared to controls [58] (for an extensive review on the connection between LRP6 and AD the reader is referred to [59]). Another Wnt signalling suppressing component in AD is the induced neuronal expression of the endogenous Wnt signalling antagonist DKK1 in the brains of post-mortem AD patients as well as in AD mouse models [60][61]. In fact, it was demonstrated that AD-associated Aβ fibrils induce Dkk1 expression in acute mouse hippocampal slices within several hours [62]. Importantly, DKK1 is a ligand for LRP6 and exerts it action by removing LRP6 from functional canonical Fz-LRP6 receptor heterodimers [45]. Interestingly, the Dkk1 homologue Dkk2 was also significantly upregulated in various AD mouse models, specifically within the myeloid cell lineage that includes microglia and other immune cells [63]. The protein clusterin also participates in the Aβ-DKK1 pathway. From a purely genetic standpoint, the encoding gene CLU had previously been identified as a major LOAD risk gene in genome wide association studies (GWAS), where various AD-linked single cell nucleotide polymorphisms were found to be associated with AD [64][65].
ApoE is a regulator of lipid homeostasis predominantly produced by astrocytes within the CNS. By binding to ApoE receptors on neurons, it transports cholesterol to neurons, which lack cholesterol producing capabilities. [66][67]. However, among the three existing polymorphic alleles ε2, ε3 and ε4, the APOE ε4 allele is found in ~40% of AD cases despite only being represented in 13.7% of the general population, thus classifying it as a major AD risk gene [68]. The nature of the role of ApoE in AD is multifaceted and still not fully understood, but highlighted by increased Aβ binding and deposition in APOE ε4 carriers [69]. In addition, collective evidence suggests that ApoE also interacts with Wnt signalling pathways in an AD-relevant manner: ApoE4-mediated Aβ pathology in the APP/PS1 AD mouse model required the expression of its neuronal receptor LRP1 [70], which had previously been found to suppress Wnt3a-driven canonical Wnt signalling in HEK cells by interacting with Fz1 [71]. Furthermore, ApoE4 treatment in PC12 cells, more so than ApoE2 and ApoE3, suppressed Wnt7a-stimulated canonical signalling [72].
Genetic variants of triggering receptor expressed on myeloid cells-2 (TREM2) have also been linked to increased susceptibility to LOAD [73][74]. Although relatively rare overall, carriers of the most common and best studied TREM2 variant (R47H) had a 2–4 fold increased risk for LOAD, which is in the same range as the APOE ε4 allele (minor allele frequency is population dependent but reaches up to 0.63% in the Icelandic population [73]). Within the CNS, TREM2 is predominantly expressed by microglia where it was shown to modulate canonical Wnt signalling to support microglial survival and microgliosis, both of which are markedly impaired in Trem2−/− mice [75]. These and other findings highlight the importance of the CNS immune system in AD, the proper function of which requires canonical Wnt signalling.
In addition to genetic analyses of AD pathogenic mechanisms, dynamic gene expression changes associated with AD and the resulting protein level changes are equally important for enabling our understanding of this disease. In a recent mass spectrometry based proteomics study, protein level changes were assessed in the brains and cerebrospinal fluid of AD patients and compared with control, prodromal, and mild cognitive impairment (MCI) cases [76]. Indeed, Wnt signalling related proteins were among those proteins exhibiting increased levels. These proteins included the Wnt ligands WNT5A, WNT5B as well as the endogenous antagonists SFRP1 and FRZB (SFRP3). While elevated SFRP1 and FRZB levels would be consistent with a reduced Wnt signalling tone, increased presence of WNT5A and WNT5B may reflect a pathological shift from canonical to non-canonical signalling that reduces synapse stability due to increased actin cytoskeleton dynamics [77]. In further support of a reduced Wnt signalling tone in AD brains, the phosphoproteome (phosphorylation status of proteins within the proteome) revealed significant phosphorylation increases of GSK-3β target proteins, which is indicative of increased GSK-3β activity and reduced canonical Wnt signalling strength [76]. These findings are corroborated by a further proteomics study, which reported that canonical Wnt signalling was dysregulated in specific human AD brain regions versus control [78]. The hippocampus for instance exhibited 17 differentially expressed Wnt signalling related proteins, including GSK-3β, GSK-3α, and AKT3, which were all downregulated, as well as the DKK homologue DKK3, which was upregulated. While the role of DKK3 as an agonist or antagonist of Wnt signalling is context dependent [79][80], the downregulation of GSK-3β, GSK-3α, and AKT3 would be expected to result in increased β-catenin stability and hence canonical Wnt signalling. This would be incompatible with the widespread notion that a reduction of canonical Wnt signalling contributes to AD pathology.
These findings notwithstanding, Wnt signalling dysregulation has been found to be a prominent feature in AD and further studies are needed to fully understand all the aspects involved. (Figure 2).
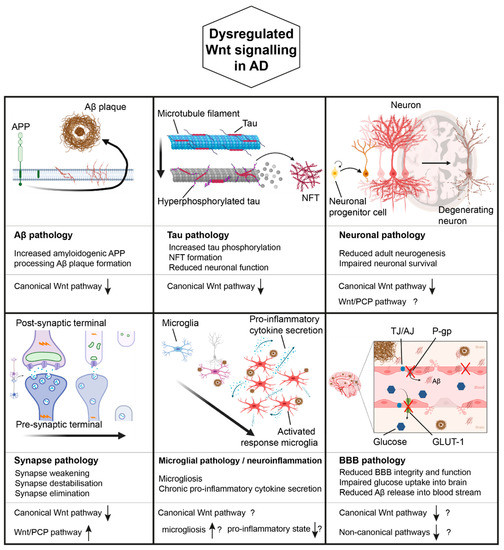
Figure 2. Wnt signalling in AD. Dysregulated Wnt signalling may contribute to various pathological manifestation of AD, including Aβ pathology, Tau pathology, neuronal pathology, synapse pathology, microglial pathology/neuroinflammation, as well as BBB pathology. Known or hypothesised (indicated by ‘?') changes in Wnt signalling associated with each manifestation are indicated (based on published literature). ↑ and ↓ indicate increased and decreased Wnt signalling respectively. Created with Biorender.com.
References
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279, doi:10.1016/j.jalz.2011.03.008.
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269, doi:10.1016/j.jalz.2011.03.005.
- Alzheimer’s Association 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387, doi:10.1016/j.jalz.2019.01.010.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608, doi:10.15252/emmm.201606210.
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New approaches for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2019, 29, 125–133, doi:10.1016/j.bmcl.2018.11.034.
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339, doi:10.1016/j.cell.2019.09.001.
- Cao, J.; Hou, J.; Ping, J.; Cai, D. Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–20, doi:10.1186/s13024-018-0299-8.
- Vermunt, L.; Sikkes, S.A.; Hout, A.V.D.; Handels, R.; Bos, I.; Van Der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898, doi:10.1016/j.jalz.2019.04.001.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562, doi:10.1016/j.jalz.2018.02.018.
- Sayas, C.L. Tau-based therapies for Alzheimer’s disease: Promising novel neuroprotective approaches. In Neuroprotection in Autism, Schizophrenia and Alzheimer’s Disease; Elsevier BV, 2020; pp. 245–272.
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227, doi:10.3389/fncel.2019.00227.
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74, doi:10.1093/jmcb/mjt051.
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nat. Cell Biol. 2003, 423, 448–452, doi:10.1038/nature01611.
- Logan, C.Y.; Nusse, R. THE WNT SIGNALING PATHWAY IN DEVELOPMENT AND DISEASE. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810, doi:10.1146/annurev.cellbio.20.010403.113126.
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, doi:10.1016/j.cell.2012.05.012.
- Schulte, G.; Bryja, V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 518–525, doi:10.1016/j.tips.2007.09.001.
- Macdonald, B.T.; He, X. Frizzled and LRP5/6 Receptors for Wnt/ -Catenin Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880, doi:10.1101/cshperspect.a007880.
- Fradkin, L.G.; Dura, J.-M.; Noordermeer, J.N. Ryks: new partners for Wnts in the developing and regenerating nervous system. Trends Neurosci. 2010, 33, 84–92, doi:10.1016/j.tins.2009.11.005.
- Green, J.L.; Kuntz, S.G.; Sternberg, P.W. Ror receptor tyrosine kinases: orphans no more. Trends Cell Biol. 2008, 18, 536–544, doi:10.1016/j.tcb.2008.08.006.
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.-C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nat. Cell Biol. 1996, 382, 225–230, doi:10.1038/382225a0.
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural Basis of Wnt Recognition by Frizzled. Sci. 2012, 337, 59–64, doi:10.1126/science.1222879.
- Dann, C.E.; Hsieh, J.-C.; Rattner, A.; Sharma, D.; Nathans, J.; Leahy, D.J. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nat. Cell Biol. 2001, 412, 86–90, doi:10.1038/35083601.
- Kikuchi, A.; Yamamoto, H.; Sato, A.; Matsumoto, S. New Insights into the Mechanism of Wnt Signaling Pathway Activation. International Review of Cell and Molecular Biology 2011, 291, 21–71, doi:10.1016/b978-0-12-386035-4.00002-1.
- Kikuchi, A.; Yamamoto, H.; Sato, A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 2009, 19, 119–129, doi:10.1016/j.tcb.2009.01.003.
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26.
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.-H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847, doi:10.1016/s0092-8674(02)00685-2.
- Huber, O.; Korn, R.; McLaughlin, J.; Ohsugi, M.; Herrmann, B.G.; Kemler, R. Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech. Dev. 1996, 59, 3–10, doi:10.1016/0925-4773(96)00597-7.
- Molenaar, M.; Van De Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 Transcription Factor Mediates β-Catenin-Induced Axis Formation in Xenopus Embryos. Cell 1996, 86, 391–399, doi:10.1016/s0092-8674(00)80112-9.
- Vlad, A.; Röhrs, S.; Klein-Hitpass, L.; Müller, O. The first five years of the Wnt targetome. Cell. Signal. 2008, 20, 795–802, doi:10.1016/j.cellsig.2007.10.031.
- Macheda, M.L.; Sun, W.W.; Kugathasan, K.; Hogan, B.M.; Bower, N.I.; Halford, M.M.; Zhang, Y.F.; Jacques, B.E.; Lieschke, G.J.; Dabdoub, A.; et al. The Wnt Receptor Ryk Plays a Role in Mammalian Planar Cell Polarity Signaling. J. Biol. Chem. 2012, 287, 29312–29323, doi:10.1074/jbc.m112.362681.
- Nishita, M.; Itsukushima, S.; Nomachi, A.; Endo, M.; Wang, Z.; Inaba, D.; Qiao, S.; Takada, S.; Kikuchi, A.; Minami, Y. Ror2/Frizzled Complex Mediates Wnt5a-Induced AP-1 Activation by Regulating Dishevelled Polymerization. Mol. Cell. Biol. 2010, 30, 3610–3619, doi:10.1128/mcb.00177-10.
- Green, J.; Nusse, R.; Van Amerongen, R. The Role of Ryk and Ror Receptor Tyrosine Kinases in Wnt Signal Transduction. Cold Spring Harb. Perspect. Biol. 2014, 6, a009175, doi:10.1101/cshperspect.a009175.
- Strutt, D.; Weber, U.; Mlodzik, M. The role of RhoA in tissue polarity and Frizzled signalling. Nat. Cell Biol. 1997, 387, 292–295, doi:10.1038/387292a0.
- Fanto, M.; Weber, U.; Strutt, D.; Mlodzik, M. Nuclear signaling by Rac and Rho GTPases is required in the establishment of epithelial planar polarity in the Drosophila eye. Curr. Biol. 2000, 10, 979–S1, doi:10.1016/s0960-9822(00)00645-x.
- Boutros, M.; Paricio, N.; Strutt, D.I.; Mlodzik, M. Dishevelled Activates JNK and Discriminates between JNK Pathways in Planar Polarity and wingless Signaling. Cell 1998, 94, 109–118, doi:10.1016/s0092-8674(00)81226-x.
- Winter, C.G.; Wang, B.; Ballew, A.; Royou, A.; Karess, R.; Axelrod, J.D.; Luo, L. Drosophila Rho-Associated Kinase (Drok) Links Frizzled-Mediated Planar Cell Polarity Signaling to the Actin Cytoskeleton. Cell 2001, 105, 81–91, doi:10.1016/s0092-8674(01)00298-7.
- Seifert, J.R.K.; Mlodzik, M. Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat. Rev. Genet. 2007, 8, 126–138, doi:10.1038/nrg2042.
- Slusarski, D.C.; Corces, V.G.; Moon, R.T. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nat. Cell Biol. 1997, 390, 410–413, doi:10.1038/37138.
- Kühl, M.; Sheldahl, L.C.; Park, M.; Miller, J.R.; Moon, R.T. The Wnt/Ca2+ pathway A new vertebrate Wnt signaling pathway takes shape. Trends Genet. 2000, 16, 279–283.
- Slusarski, D.C.; Yang-Snyder, J.; Busa, W.B.; Moon, R.T. Modulation of Embryonic Intracellular Ca2+Signaling byWnt-5A. Dev. Biol. 1997, 182, 114–120, doi:10.1006/dbio.1996.8463.
- Sheldahl, L.C.; Park, M.; Malbon, C.C.; Moon, R.T. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in aG-protein-dependent manner. Curr. Biol. 1999, 9, 695–S1, doi:10.1016/s0960-9822(99)80310-8.
- Kühl, M.; Sheldahl, L.C.; Malbon, C.C.; Moon, R.T. Ca2+/Calmodulin-dependent Protein Kinase II Is Stimulated by Wnt and Frizzled Homologs and Promotes Ventral Cell Fates inXenopus. J. Biol. Chem. 2000, 275, 12701–12711, doi:10.1074/jbc.275.17.12701.
- De, A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim. et Biophys. Sin. 2011, 43, 745–756, doi:10.1093/abbs/gmr079.
- Cruciat, C.-M.; Niehrs, C. Secreted and Transmembrane Wnt Inhibitors and Activators. Cold Spring Harb. Perspect. Biol. 2012, 5, a015081, doi:10.1101/cshperspect.a015081.
- Semënov, M.V.; Tamai, K.; Brott, B.K.; Kühl, M.; Sokol, S.; He, X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr. Biol. 2001, 11, 951–961, doi:10.1016/s0960-9822(01)00290-1.
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B.M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; et al. Kremen proteins are Dickkopf receptors that regulate Wnt/β-catenin signalling. Nat. Cell Biol. 2002, 417, 664–667, doi:10.1038/nature756.
- Mao, B.; Niehrs, C. Kremen2 modulates Dickkopf2 activity during Wnt/lRP6 signaling. Gene 2003, 302, 179–183, doi:10.1016/s0378-1119(02)01106-x.
- Hao, H.-X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nat. Cell Biol. 2012, 485, 195–200, doi:10.1038/nature11019.
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316, doi:10.1101/gad.235473.113.
- Ohkawara, B.; Glinka, A.; Niehrs, C. Rspo3 Binds Syndecan 4 and Induces Wnt/PCP Signaling via Clathrin-Mediated Endocytosis to Promote Morphogenesis. Dev. Cell 2011, 20, 303–314, doi:10.1016/j.devcel.2011.01.006.
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular Development in the Retina and Inner Ear. Cell 2004, 116, 883–895, doi:10.1016/s0092-8674(04)00216-8.
- Oliva, C.A.; Montecinos-Oliva, C.; Inestrosa, N.C. Wnt Signaling in the Central Nervous System: New Insights in Health and Disease. In Progress in Molecular Biology and Translational Science; Elsevier BV, 2018; Vol. 153, pp. 81–130.
- Anderton, B.H.; Dayanandan, R.; Killick, R.; Lovestone, S. Does dysregulation of the Notch and wingless/Wnt pathways underlie the pathogenesis of Alzheimer’s disease? Mol. Med. Today 2000, 6, 54–59, doi:10.1016/s1357-4310(99)01640-8.
- Van Der Flier, W.M. Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 2005, 76, v2–v7, doi:10.1136/jnnp.2005.082867.
- Mateo, I.; Infante, J.; Llorca, J.; Rodríguez, E.; Berciano, J.; Combarros, O. Association between Glycogen Synthase Kinase-3β Genetic Polymorphism and Late-Onset Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2006, 21, 228–232, doi:10.1159/000091044.
- De Ferrari, G.V.; Papassotiropoulos, A.; Biechele, T.; De-Vrieze, F.W.; Avila, M.E.; Major, M.B.; Myers, A.; Sáez, K.; Henríquez, J.P.; Zhao, A.; et al. Common genetic variation within the Low-Density Lipoprotein Receptor-Related Protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. 2007, 104, 9434–9439, doi:10.1073/pnas.0603523104.
- Alarcón, M.A.; Medina, M.A.; Hu, Q.; Avila, M.E.; Bustos, B.I.; Pérez-Palma, E.; Peralta, A.; Salazar, P.; Ugarte, G.D.; Reyes, A.E.; et al. A novel functional low-density lipoprotein receptor-related protein 6 gene alternative splice variant is associated with Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1709.e9–1709.e18, doi:10.1016/j.neurobiolaging.2012.11.004.
- Liu, C.-C.; Tsai, C.-W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H.; et al. Deficiency in LRP6-Mediated Wnt Signaling Contributes to Synaptic Abnormalities and Amyloid Pathology in Alzheimer’s Disease. Neuron 2014, 84, 63–77, doi:10.1016/j.neuron.2014.08.048.
- Buechler, J.; Salinas, P.C. Deficient Wnt Signaling and Synaptic Vulnerability in Alzheimer’s Disease: Emerging Roles for the LRP6 Receptor. Front. Synaptic Neurosci. 2018, 10, 38,, doi:10.3389/fnsyn.2018.00038.
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a Negative Modulator of the Wnt Pathway, Is Associated with Neuronal Degeneration in Alzheimer’s Brain. J. Neurosci. 2004, 24, 6021–6027, doi:10.1523/jneurosci.1381-04.2004.
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551, doi:10.1111/j.1471-4159.2009.06566.x.
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The Secreted Wnt Antagonist Dickkopf-1 Is Required for Amyloid -Mediated Synaptic Loss. J. Neurosci. 2012, 32, 3492–3498, doi:10.1523/jneurosci.4562-11.2012.
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847, doi:10.1016/j.celrep.2017.12.066.
- Lambert, J.-C.; the European Alzheimer’s Disease Initiative Investigators; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; O Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099, doi:10.1038/ng.439.
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093, doi:10.1038/ng.440.
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118, doi:10.1038/nrneurol.2012.263.
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344, doi:10.1038/nrn2620.
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.T.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease. JAMA 1997, 278, 1349–1356, doi:10.1001/jama.1997.03550160069041.
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. 1993, 90, 9649–9653, doi:10.1073/pnas.90.20.9649.
- Tachibana, M.; Holm, M.-L.; Liu, C.-C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277, doi:10.1172/jci124853.
- Zilberberg, A.; Yaniv, A.; Gazit, A. The Low Density Lipoprotein Receptor-1, LRP1, Interacts with the Human Frizzled-1 (HFz1) and Down-regulates the Canonical Wnt Signaling Pathway. J. Biol. Chem. 2004, 279, 17535–17542, doi:10.1074/jbc.m311292200.
- Caruso, A.; Motolese, M.; Iacovelli, L.; Caraci, F.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; Gaviraghi, G.; Caricasole, A. Inhibition of the canonical Wnt signaling pathway by apolipoprotein E4 in PC12 cells. J. Neurochem. 2006, 98, 364–371, doi:10.1111/j.1471-4159.2006.03867.x.
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher-Moser, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. New Engl. J. Med. 2013, 368, 107–116, doi:10.1056/nejmoa1211103.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.G.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127, doi:10.1056/nejmoa1211851.
- Zheng, H.; Jia, L.; Liu, C.-C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.-F.; Fryer, J.D.; Wang, X.; Zhang, Y.-W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784, doi:10.1523/jneurosci.2459-16.2017.
- Bai, B.; Wang, X.; Li, Y.; Chen, P.-C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991.e7, doi:10.1016/j.neuron.2019.12.015.
- Elliott, C.; Rojo, A.I.; Ribe, E.; Broadstock, M.; Xia, W.; Morin, P.; Semenov, M.; Baillie, G.; Cuadrado, A.; Al-Shawi, R.; et al. A role for APP in Wnt signalling links synapse loss with β-amyloid production. Transl. Psychiatry 2018, 8, 1–13, doi:10.1038/s41398-018-0231-6.
- Xu, J.; Patassini, S.; Rustogi, N.; Riba-Garcia, I.; Hale, B.D.; Phillips, A.M.; Waldvogel, H.; Haines, R.; Bradbury, P.; Stevens, A.; et al. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun. Biol. 2019, 2, 1–15, doi:10.1038/s42003-018-0254-9.
- Nakamura, R.E.; Hunter, D.D.; Yi, H.; Brunken, W.J.; Hackam, A.S. Identification of two novel activities of the Wnt signaling regulator Dickkopf 3 and characterization of its expression in the mouse retina. BMC Cell Biol. 2007, 8, 52, doi:10.1186/1471-2121-8-52.
- Yue, W.; Sun, Q.; Dacic, S.; Landreneau, R.J.; Siegfried, J.M.; Yu, J.; Zhang, L. Downregulation of Dkk3 activates β-catenin/TCF-4 signaling in lung cancer. Carcinog. 2008, 29, 84–92, doi:10.1093/carcin/bgm267.


