 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Arjun Patel | + 1710 word(s) | 1710 | 2021-01-06 06:59:55 | | | |
| 2 | Arjun Patel | Meta information modification | 1710 | 2021-01-14 22:29:24 | | | | |
| 3 | Vicky Zhou | Meta information modification | 1710 | 2021-01-15 03:11:05 | | |
Video Upload Options
Microbial strains are being engineered for an increasingly diverse array of applications, from chemical production to human health. While traditional engineering disciplines are driven by predictive design tools, these tools have been difficult to build for biological design due to the complexity of biological systems and many unknowns of their quantitative behavior. However, due to many recent advances, the gap between design in biology and other engineering fields is closing.
1. Introduction
Microbes have been engineered for a broad number of applications. As cell factories, cells have been designed to convert low-value substrates into valuable chemical products, including biofuels [1], commodity chemicals [2], bioactive compounds [3], and foods [4]. To benefit the environment, microbes have been engineered for bioremediation [5] and biosensing [6] of toxic compounds and pollutants. As engineered tools, microbes have been programmed using cell circuits to exhibit elaborate behaviors, from synchronized fluorescence [7] to hunting down tumors to deliver chemotherapeutics [8]. Finally, as cellular products, microbes themselves are increasingly of interest for probiotic and nutritional supplements [9].
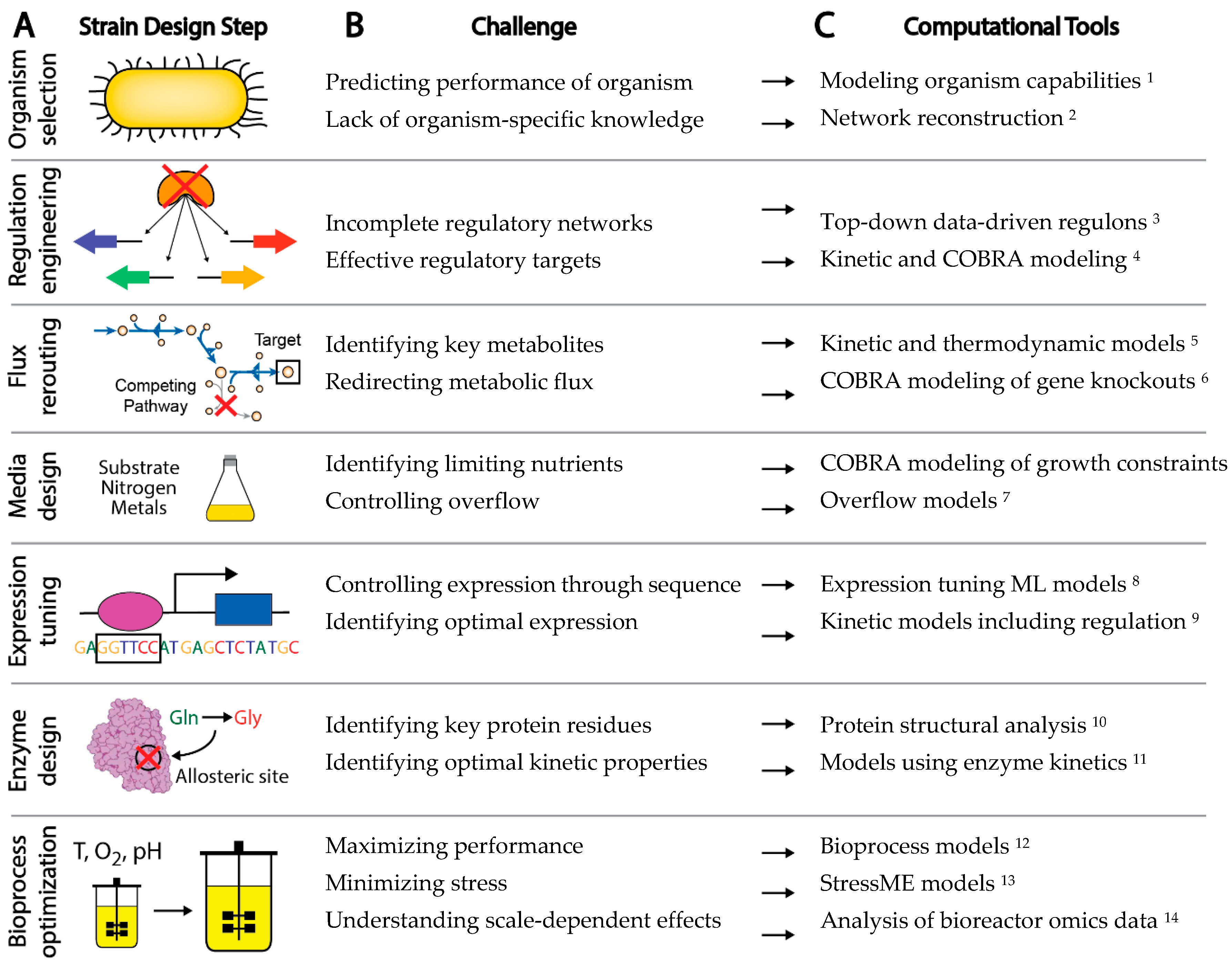
The experimental workflow to engineer a new microbial strain has a number of common steps, although the order may vary (Figure 1A) [10]. First, a background organism and strain is chosen for the application of interest. Genes may be knocked out, introduced, knocked down, or overexpressed for a variety of purposes, such as control of transcriptional regulation, redirection of metabolic flux to desired pathways, or removal of unwanted or wasteful processes. Bioprocess conditions can be optimized through control of various factors including media, feed rate, growth rate, pH, and temperature. Specific sequence variants can be introduced through rational design or selected through screens and adaptive laboratory evolution to control expression, alter enzyme activity, or remove regulatory sites from proteins. The typical strain design workflow thus requires a large number of decisions on how to improve strain behavior. Left to a trial and error approach, the complexity of biological systems makes efficient engineering of strains a daunting task.
2. Computational Tools
To aid strain design efforts, computational tools have been integrated from various fields into the strain design workflow [27]. These tools offer the promise of restricting the experimental search space by either identifying modifications that are more likely to improve strain performance or proposing entirely new designs through mathematical modeling of cell behavior. However, many steps in the strain design process are still driven by rational approaches, rules of thumb, and extensive experimental screening and trial and error. Workflows driven purely by predictive tools would have the advantage of efficiency of execution through fewer experimental steps, reduced time, and ultimately improved performance through careful guidance toward an optimal desired phenotype. We describe two approaches that show promise as systematic tools for cell design: genetic circuits and genome-scale modeling.
One strategy for constructing synthetic strains has been to engineer desired behaviors through the use of genetic circuits [28]. The key concept is to carefully characterize and often mathematically model the behavior of a ‘circuit’, typically a small transcriptional regulatory network, to control a cell phenotype. As greater numbers of these small circuits are characterized, they begin to comprise a ‘parts list’ of available phenotypes from which an engineer can choose or can be assembled automatically by an algorithm [29]. Larger and larger circuits can then be constructed of well-characterized smaller circuits to engineer more complex phenotypes. This strategy has been employed for a number of promising applications [30][31].
Another successful paradigm for computational design of cells is genome-scale network modeling [32]. While genetic circuits approaches utilize highly controllable systems of limited scope, genome-scale models seek to predict cell phenotype by comprehensively modeling all known functions of the cell. As part of the Constraint-based Reconstruction and Analysis (COBRA) framework, genome-scale models of metabolism utilize a metabolic network reconstruction to predict metabolic phenotypes and analyze genome-scale datasets [33]. These models deal with the large scope of the system by utilizing the constraint-based modeling framework, which requires few parameters to generate predictions. The challenge of managing these large-scale models is achieved through community enforcement of rigid requirements, testing, and data standards [12][34]. Although these models were originally developed for metabolism, they have recently been extended to include transcription and translation machinery [35][36][37] and even further to whole-cell kinetic simulations [38].
Although computational methods have undoubtedly augmented rational strain design efforts, there are a number of challenges in a strain design workflow that still cannot be effectively addressed by existing computational tools [39] (Figure 1B). For example: (1) Organisms are often chosen for a strain design project due to historical knowledge and convenience, rather than fundamental benefits provided by the organism that could be calculated computationally a priori, (2) Gaps in gene annotation make choosing non-model organisms a risk, (3) The difficulty in accounting for enzyme kinetics makes the understanding of metabolic and allosteric regulation a challenge, (4) A lack of understanding of regulatory networks impedes the understanding and control of gene expression, and (5) Insufficient annotation of the organism genome makes it difficult to interpret the functional implications of sequence variation. Challenges such as these present major barriers to interpreting data and predicting strain phenotype.
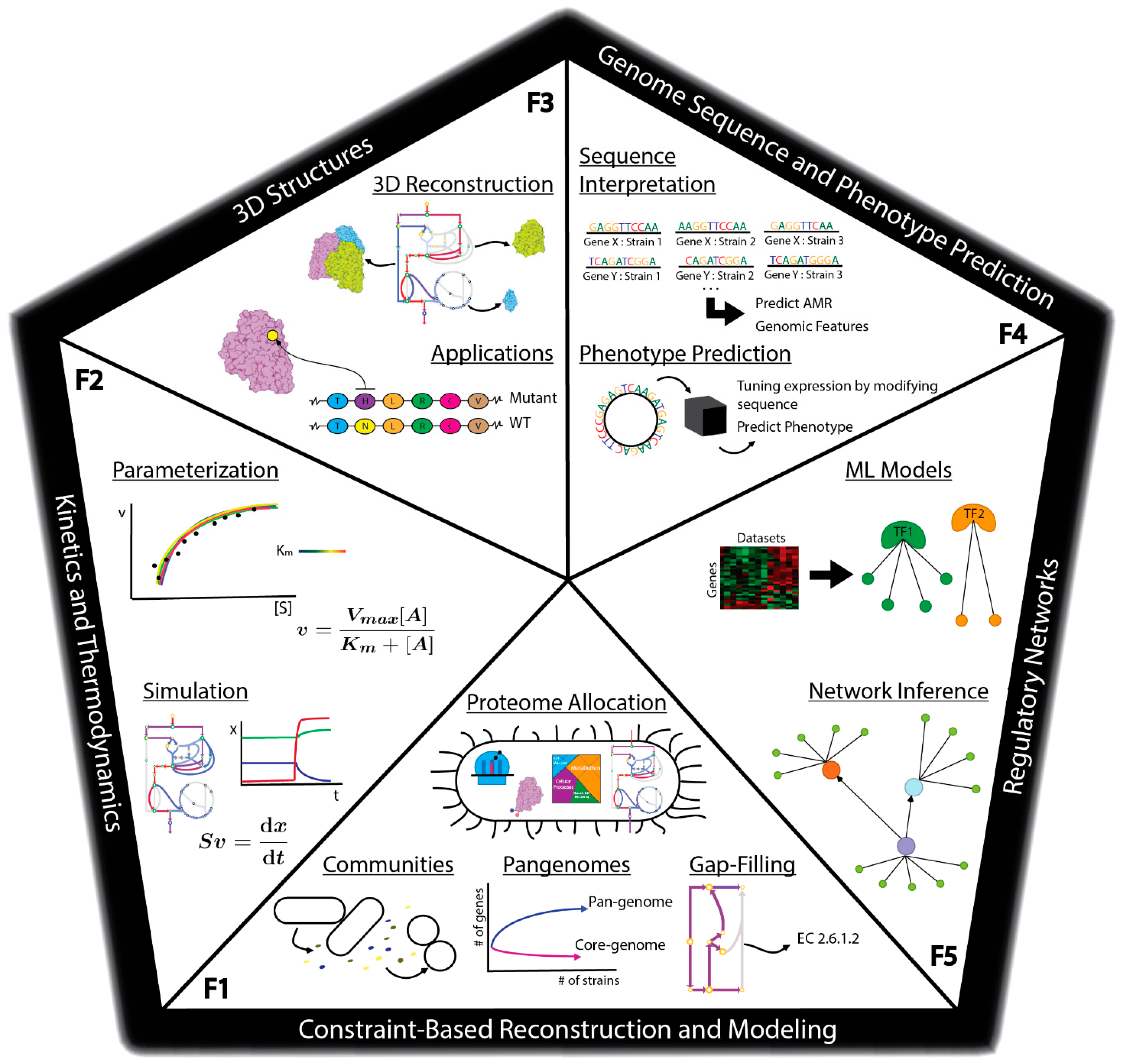
There are many methods currently being developed that may directly meet these challenges to enable fully predictive strain design workflows (Figure 1C). For example, advances in metabolic modeling could enable the optimization of bioprocess conditions or the identification of optimal expression levels of pathway genes [40][14]. However, these models are still in development and have not yet been shown to enable accurate predictions at scale. In this perspective, we describe five frontiers consisting of promising developments in computational strain design that may pave the way toward achieving comprehensive and integrated strain design workflows (Figure 2).
3. Outlook for Synthetic Genome Design
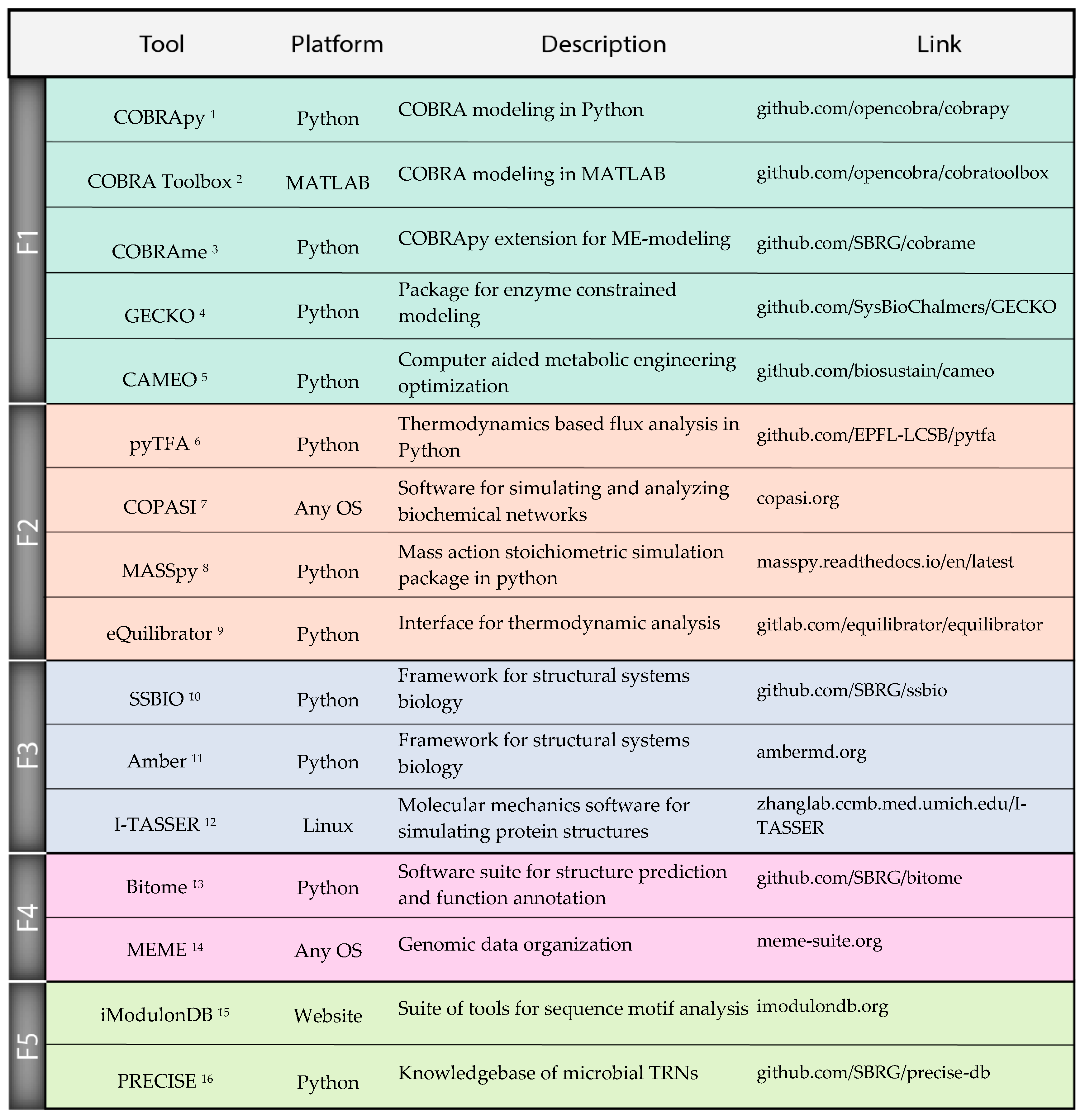
Although there has been substantial progress in each of the individual fields discussed above, there are additional challenges with integrating these tools into an effective strain design workflow. Workflow: While we discussed many tools as they relate to individual strain design tasks, these tasks must be synthesized into a coherent end-to-end design workflow. The decisions of the order of operations in the development of a strain could greatly benefit from computational predictions, but much work is yet to be done to identify a strain design workflow that maximizes efficiency and minimizes cost and risk. Expertise: Any workflow that integrates many different computational tools will require domain expertise in each tool to decide details of implementation, from parameters to valid use cases. Thus, strain designers will be required to have broad computational skillsets that exceed what is taught by most current training programs. Software: The practical difficulty of implementing many separate computational tools can become a substantial burden, spanning various details from licensing issues to file formats. However, the number of software packages enabling these workflows continues to increase, and we mention many examples in this work (Figure 3). Thanks to these efforts, finding compatible tools for easily integrated workflows is becoming easier. Validation: Tools must be validated to clearly established accuracy metrics under physiological conditions. Validation of tools on individual datasets, for example on a single wild type strain background, is likely to be insufficient as the strain is engineered further from the wild type. To meet these challenges, it is critical to take a systematic approach that includes dedicated training, effective documentation of tools, and extensive validation of tools in real applications. There will be a significant challenge reaching a standard where strain design researchers can effectively conduct analyses and understand results from multiple tools across a typical workflow.
The field is nearing an important milestone in synthetic biology, that of the comprehensive and computationally-driven strain design workflow. We may soon enter an era of ‘computational genome design’, where rational approaches finally give way to biological design algorithms dominated by computational predictions. Thus, one of the early promises of the field of systems biology may finally be nearing its realization. The practical applications of such a cell design workflow are endless, from the chemical industry to the environment to human health.
References
- Liao, J.C.; Mi, L.; Pontrelli, S.; Luo, S. Fuelling the Future: Microbial Engineering for the Production of Sustainable Biofuels. Nat. Rev. Microbiol. 2016, 14, 288–304.
- Lee, S.Y.; Kim, H.U.; Chae, T.U.; Cho, J.S.; Kim, J.W.; Shin, J.H.; Kim, D.I.; Ko, Y.-S.; Jang, W.D.; Jang, Y.-S. A Comprehensive Metabolic Map for Production of Bio-Based Chemicals. Nat. Catal. 2019, 2, 18–33.
- Kalia, V.C.; Saini, A.K. (Eds.) Metabolic Engineering for Bioactive Compounds: Strategies and Processes; Springer: Singapore, 2017.
- Matassa, S.; Boon, N.; Pikaar, I.; Verstraete, W. Microbial Protein: Future Sustainable Food Supply Route with Low Environmental Footprint. Microb. Biotechnol. 2016, 9, 568–575.
- Das, S.; Dash, H.R. 1—Microbial Bioremediation: A Potential Tool for Restoration of Contaminated Areas. In Microbial Biodegradation and Bioremediation; Das, S., Ed.; Elsevier: Oxford, UK, 2014; pp. 1–21.
- Bereza-Malcolm, L.T.; Mann, G.; Franks, A.E. Environmental Sensing of Heavy Metals Through Whole Cell Microbial Biosensors: A Synthetic Biology Approach. ACS Synth. Biol. 2015, 4, 535–546.
- Danino, T.; Mondragón-Palomino, O.; Tsimring, L.; Hasty, J. A Synchronized Quorum of Genetic Clocks. Nature 2010, 463, 326–330.
- Din, M.O.; Danino, T.; Prindle, A.; Skalak, M.; Selimkhanov, J.; Allen, K.; Julio, E.; Atolia, E.; Tsimring, L.S.; Bhatia, S.N.; et al. Synchronized Cycles of Bacterial Lysis for in Vivo Delivery. Nature 2016, 536, 81–85.
- Yadav, R.; Singh, P.K.; Shukla, P. Metabolic Engineering for Probiotics and Their Genome-Wide Expression Profiling. Curr. Protein Pept. Sci. 2018, 19, 68–74.
- Lee, S.Y.; Kim, H.U. Systems Strategies for Developing Industrial Microbial Strains. Nat. Biotechnol. 2015, 33, 1061–1072.
- Monk, J.M.; Koza, A.; Campodonico, M.A.; Machado, D.; Seoane, J.M.; Palsson, B.O.; Herrgård, M.J.; Feist, A.M. Multi-Omics Quantification of Species Variation of Escherichia Coli Links Molecular Features with Strain Phenotypes. Cell Syst. 2016, 3, 238–251.e12.
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A Systematic Assessment of Current Genome-Scale Metabolic Reconstruction Tools. Genome Biol. 2019, 20, 158.
- Sastry, A.V.; Gao, Y.; Szubin, R.; Hefner, Y.; Xu, S.; Kim, D.; Choudhary, K.S.; Yang, L.; King, Z.A.; Palsson, B.O. The Escherichia Coli Transcriptome Mostly Consists of Independently Regulated Modules. Nat. Commun. 2019, 10, 5536.
- Andreozzi, S.; Chakrabarti, A.; Soh, K.C.; Burgard, A.; Yang, T.H.; Van Dien, S.; Miskovic, L.; Hatzimanikatis, V. Identification of Metabolic Engineering Targets for the Enhancement of 1,4-Butanediol Production in Recombinant E. Coli Using Large-Scale Kinetic Models. Metab. Eng. 2016, 35, 148–159.
- Kümmel, A.; Panke, S.; Heinemann, M. Putative Regulatory Sites Unraveled by Network-Embedded Thermodynamic Analysis of Metabolome Data. Mol. Syst. Biol. 2006, 2, 2006.0034.
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A Bilevel Programming Framework for Identifying Gene Knockout Strategies for Microbial Strain Optimization. Biotechnol. Bioeng. 2003, 84, 647–657.
- De Groot, D.H.; Lischke, J.; Muolo, R.; Planqué, R.; Bruggeman, F.J.; Teusink, B. The Common Message of Constraint-Based Optimization Approaches: Overflow Metabolism Is Caused by Two Growth-Limiting Constraints. Cell. Mol. Life Sci. 2020, 77, 441–453.
- Zrimec, J.; Börlin, C.S.; Buric, F.; Muhammad, A.S.; Chen, R.; Siewers, V.; Verendel, V.; Nielsen, J.; Töpel, M.; Zelezniak, A. Deep Learning Suggests That Gene Expression Is Encoded in All Parts of a Co-Evolving Interacting Gene Regulatory Structure. Nat. Commun. 2020, 11, 6141.
- Kotte, O.; Zaugg, J.B.; Heinemann, M. Bacterial Adaptation through Distributed Sensing of Metabolic Fluxes. Mol. Syst. Biol. 2010, 6, 355.
- Brunk, E.; Mih, N.; Monk, J.; Zhang, Z.; O’Brien, E.J.; Bliven, S.E.; Chen, K.; Chang, R.L.; Bourne, P.E.; Palsson, B.O. Systems Biology of the Structural Proteome. BMC Syst. Biol. 2016, 10, 1–6.
- Kim, O.D.; Rocha, M.; Maia, P. A Review of Dynamic Modeling Approaches and Their Application in Computational Strain Optimization for Metabolic Engineering. Front. Microbiol. 2018, 9, 1690.
- Jabarivelisdeh, B.; Waldherr, S. Optimization of Bioprocess Productivity Based on Metabolic-Genetic Network Models with Bilevel Dynamic Programming. Biotechnol. Bioeng. 2018, 115, 1829–1841.
- Chen, K.; Gao, Y.; Mih, N.; O’Brien, E.J.; Yang, L.; Palsson, B.O. Thermosensitivity of Growth Is Determined by Chaperone-Mediated Proteome Reallocation. Proc. Natl. Acad. Sci. USA 2017, 114, 11548–11553.
- Du, B.; Yang, L.; Lloyd, C.J.; Fang, X.; Palsson, B.O. Genome-Scale Model of Metabolism and Gene Expression Provides a Multi-Scale Description of Acid Stress Responses in Escherichia Coli. PLoS Comput. Biol. 2019, 15, e1007525.
- Yang, L.; Mih, N.; Anand, A.; Park, J.H.; Tan, J.; Yurkovich, J.T.; Monk, J.M.; Lloyd, C.J.; Sandberg, T.E.; Seo, S.W.; et al. Cellular Responses to Reactive Oxygen Species Are Predicted from Molecular Mechanisms. Proc. Natl. Acad. Sci. USA 2019, 116, 14368–14373.
- Wang, G.; Haringa, C.; Tang, W.; Noorman, H.; Chu, J.; Zhuang, Y.; Zhang, S. Coupled Metabolic-Hydrodynamic Modeling Enabling Rational Scale-up of Industrial Bioprocesses. Biotechnol. Bioeng. 2020, 117, 844–867.
- St. John, P.C.; Bomble, Y.J. Approaches to Computational Strain Design in the Multiomics Era. Front. Microbiol. 2019, 10.
- Brophy, J.A.N.; Voigt, C.A. Principles of Genetic Circuit Design. Nat. Methods 2014, 11, 508–520.
- Nielsen, A.A.K.; Der, B.S.; Shin, J.; Vaidyanathan, P.; Paralanov, V.; Strychalski, E.A.; Ross, D.; Densmore, D.; Voigt, C.A. Genetic Circuit Design Automation. Science 2016, 352, aac7341.
- Sedlmayer, F.; Aubel, D.; Fussenegger, M. Synthetic Gene Circuits for the Detection, Elimination and Prevention of Disease. Nat. Biomed. Eng. 2018, 2, 399–415.
- Khalil, A.S.; Collins, J.J. Synthetic Biology: Applications Come of Age. Nat. Rev. Genet. 2010, 11, 367–379.
- Kim, W.J.; Kim, H.U.; Lee, S.Y. Current State and Applications of Microbial Genome-Scale Metabolic Models. Curr. Opin. Syst. Biol. 2017, 2, 10–18.
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and Analysis of Biochemical Constraint-Based Models Using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702.
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for Standardized Genome-Scale Metabolic Model Testing. Nat. Biotechnol. 2020, 38, 272–276.
- Lloyd, C.J.; Ebrahim, A.; Yang, L.; King, Z.A.; Catoiu, E.; O’Brien, E.J.; Liu, J.K.; Palsson, B.O. COBRAme: A Computational Framework for Genome-Scale Models of Metabolism and Gene Expression. PLoS Comput. Biol. 2018, 14, e1006302.
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using Genome-Scale Models to Predict Biological Capabilities. Cell 2015, 161, 971–987.
- O’Brien, E.J.; Lerman, J.A.; Chang, R.L.; Hyduke, D.R.; Palsson, B.Ø. Genome-Scale Models of Metabolism and Gene Expression Extend and Refine Growth Phenotype Prediction. Mol. Syst. Biol. 2013, 9, 693.
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B., Jr.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A Whole-Cell Computational Model Predicts Phenotype from Genotype. Cell 2012, 150, 389–401.
- McCloskey, D.; Palsson, B.Ø.; Feist, A.M. Basic and Applied Uses of Genome-Scale Metabolic Network Reconstructions of Escherichia Coli. Mol. Syst. Biol. 2013, 9, 661.
- Richelle, A.; David, B.; Demaegd, D.; Dewerchin, M.; Kinet, R.; Morreale, A.; Portela, R.; Zune, Q.; von Stosch, M. Towards a Widespread Adoption of Metabolic Modeling Tools in Biopharmaceutical Industry: A Process Systems Biology Engineering Perspective. NPJ Syst. Biol. Appl. 2020, 6, 6.
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74.
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.-J.; Kerkhoven, E.J.; Nielsen, J. Improving the Phenotype Predictions of a Yeast Genome-Scale Metabolic Model by Incorporating Enzymatic Constraints. Mol. Syst. Biol. 2017, 13, 935.
- Cardoso, J.G.R.; Jensen, K.; Lieven, C.; Lærke Hansen, A.S.; Galkina, S.; Beber, M.; Özdemir, E.; Herrgård, M.J.; Redestig, H.; Sonnenschein, N. Cameo: A Python Library for Computer Aided Metabolic Engineering and Optimization of Cell Factories. ACS Synth. Biol. 2018, 7, 1163–1166.
- Salvy, P.; Fengos, G.; Ataman, M.; Pathier, T.; Soh, K.C.; Hatzimanikatis, V. pyTFA and matTFA: A Python Package and a Matlab Toolbox for Thermodynamics-Based Flux Analysis. Bioinformatics 2019, 35, 167–169.
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074.
- Haiman, Z.B.; Zielinski, D.C.; Koike, Y.; Yurkovich, J.T.; Palsson, B.O. MASSpy: Building, Simulating, and Visualizing Dynamic Biological Models in Python Using Mass Action Kinetics. bioRxiv 2020.
- Flamholz, A.; Noor, E.; Bar-Even, A.; Milo, R. eQuilibrator—the Biochemical Thermodynamics Calculator. Nucleic Acids Res. 2012, 40, D770–D775.
- Mih, N.; Brunk, E.; Chen, K.; Catoiu, E.; Sastry, A.; Kavvas, E.; Monk, J.M.; Zhang, Z.; Palsson, B.O. Ssbio: A Python Framework for Structural Systems Biology. Bioinformatics 2018, 34, 2155–2157.
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020.
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8.
- Lamoureux, C.R.; Choudhary, K.S.; King, Z.A.; Sandberg, T.E.; Gao, Y.; Sastry, A.V.; Phaneuf, P.V.; Choe, D.; Cho, B.-K.; Palsson, B.O. The Bitome: Digitized Genomic Features Reveal Fundamental Genome Organization. Nucleic Acids Res. 2020, 48, 10157–10163.
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Res. 2009, 37, W202–W208.
- Rychel, K.; Decker, K.; Sastry, A.V.; Phaneuf, P.V.; Poudel, S.; Palsson, B.O. iModulonDB: A Knowledgebase of Microbial Transcriptional Regulation Derived from Machine Learning. Nucleic Acids Res. 2020.


