 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Viktoriya Sharova | + 2477 word(s) | 2477 | 2020-12-28 09:57:28 | | | |
| 2 | Viktoriya Sharova | Meta information modification | 2477 | 2021-01-07 23:37:08 | | | | |
| 3 | Karina Chen | Meta information modification | 2477 | 2021-01-08 04:40:46 | | | | |
| 4 | Viktoriya Sharova | Meta information modification | 2477 | 2021-01-08 09:07:28 | | |
Video Upload Options
The signals generated by the HPG axis, the main participants of which are gonadotropin-releasing hormone (GnRH), gonadotropins, and sex steroids, coordinate the development and functioning of the immune system, and immunomediators, in particular, cytokines and thymic peptides, influence the HPG axis.
1. Introduction
The mechanism of GnRH secretion regulation in the hypothalamus includes a network of various neurons, including KISS1-producing ones, which can act on GnRH neurons through separate or multiple neuronal systems [1][2]. GnRH- and KISS1 neurons are located in the same regions of the hypothalamus, and GnRH neurons express KISS1R. Axons of KISS1 neurons form pericapillary plexuses at the site of GnRH secretion [3]. Neurokinin B (NKB) and dynorphin, which colocalize with KISS1 in the arcuate nucleus and are linked by axosomatic synapses, are also involved in the generation of GnRH impulses [4]. It is assumed that NKB initiates the onset of GnRH impulse secretion, while dynorphin initiates its termination [5]. General cues of GnRH neuron regulation in HPG axis is presented on Figure 1.
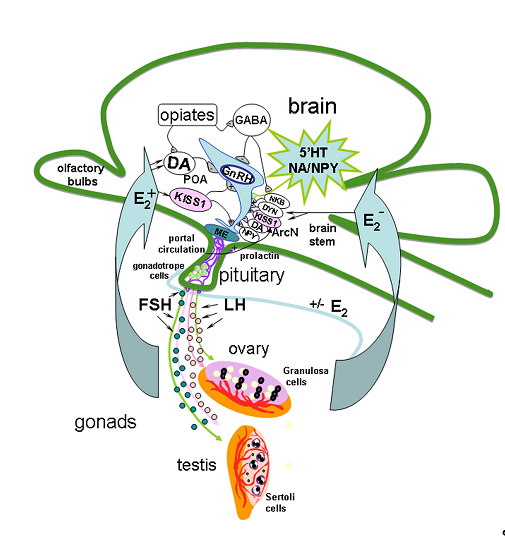
Figure 1. Model of the gonadotropin-releasing hormone (GnRH) neuron regulation in hypothalamic-pituitary-gonadal axis in adults. The mechanism of GnRH secretion regulation in the hypothalamus includes different signal molecules produced in different brain regions such as preoptic area (POA), locus coeruleus, raphe nucleus and brain stem, in particularly, serotonin (5′HT), dopamine (DА), noradrenaline (NA), gamma-aminobutyric acid (GABA), kisspeptin (KISS1), neuropeptide Y (NPY), opiates, neurokinin B (NKB) and dynorphin (DYN), which colocalize with KISS1 in the arcuate nucleus (ArcN), are also involved in the generation of GnRH impulses. NKB initiates the onset of GnRH impulse secretion in median eminence (ME), and DYN initiates its termination. In turn, axons of GnRH neurons release GnRH into the portal system in necessary concentration to trigger luteinizing hormone (LH) and follicle-stimulating hormone (FSH) secretion, which stimulate the secretion of sex steroids. Sex steroids, in turn, regulate the GnRH and LH synthesis in the brain and pituitary.
2. Effects of Different Signal Molecules on the Normal Development of the GnRH System
In vertebrates, most of the GnRH neurons are formed in the prenatal period outside the brain from the epithelium of the olfactory placodes and regulatory or morphogenetic factors are involved in the differentiation of these placodes [6]. Then GnRH neurons migrate to the forebrain, where they are also located in adults. Signaling molecules affecting the development of GnRH neurons are usually divided into groups according to functions closely related to the sites of origin, migration, and definitive location of neurons. At least five groups of signaling molecules are distinguished: (1) cell adhesion molecules, (2) soluble guidance-cue factors, (3) cytokines, (4) neurotransmitters, and (5) transcription factors (Figure 2).
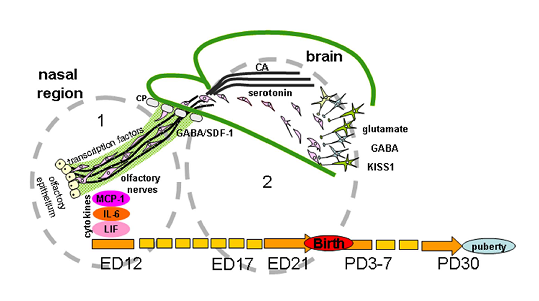
Figure 2. Regulation of gonadotropin-releasing hormone (GnRH) neuron migration in nasal region. (1) and brain (2) during development. GnRH neurons originate outside the brain in nasal olfactory epithelium and later neurons migrate using the surface of olfactory/terminal/vomeronasal nerves through the cribriform plate of the ethmoid bone (CP) to the forebrain on embryonic days (ED) 11-15 in mice and ED 12-17 in rats. Their migration is orchestrated by different signal molecules, such as cell adhesion molecules (polysialylated neural cell adhesion molecules (NCAM) in rats or peripherin in mice), soluble guidance-cue factors (semaphorins, Slit proteins, netrins, reelin, stromal cell-derived factor 1 (SDF-1), etc.), cytokines (monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6), leukemia inhibitory factor (LIF)), neurotransmitters (gamma-aminobutyric acid (GABA), glutamate, monoamines (serotonin, catecholamines (CA)), and transcription factors (Pax6, AP-2α, Gli3, Ebf2 Nhlh2 VAX1 Mash-1, Math4A, Math4/neurogenin1, NeuroD, Olf-1, GATA-4). In rats, GnRH-neurons reach their final position on ED19 and after the receiving of correct afferent innervation they are capable of performing main function—to regulate the reproductive axis in adults. The same signal molecules can perform different functions or other molecules can be included at different stages of the development of the GnRH system. For instance, NCAM apparently participate in laying the route for GnRH neurons and do not play a key role in their migration. Transcription factors are involved in the initial stages of the differentiation of olfactory placodes and the precursors of GnRH neurons, cytokines in the development of olfactory structures and in the migration of GnRH neurons and GnRH secretion. Neurotransmitters take part in the migration of GnRH neurons mainly at the stage of their entry into the forebrain. Guidance-cue molecules are involved in the development of the olfactory system and contribute to directing the migration of GnRH neurons in the nasal head region and forebrain. Kisspeptin (KISS1) signaling is well demonstrated as a key component for the onset of puberty, GnRH neurons express KISS1 in embryonic mice brain. Moreover, KISS1 may inhibit GnRH neuronal movement and plays a role of stop molecule for GnRH neuronal migration. Abbreviation: PD—postnatal day.
As noted above, GnRH neurons migrating in the nasal head region move along the olfactory, terminal, and vomeronasal nerves. After entering the forebrain, these neurons migrate caudally along a temporary projection of the vomeronasal nerve to the septopreoptic area [7]. The general pattern of GnRH system development is similar in most mammals, although there are natural differences in the timing of neuron formation and migration depending on the duration of pregnancy and the relative degree of offspring development at birth in individual species.
The process of migration is conventionally divided into three stages: (1) intranasal migration, (2) passage through the cribriform plate of the ethmoid bone, and (3) intracerebral migration (Figure 2). In normal development, each stage is characterized by a unique set of factors such as cell adhesion proteins, gradients of guidance-cue molecules, and a specific cellular microenvironment producing neurotransmitters and neuromodulators necessary for a successful migration of GnRH neurons. For example, suppression of β1-integrin, which mediates the functions of cell adhesion proteins and guidance cues, impairs the migration of GnRH neurons in mouse fetuses, with a consequent delay in puberty and impairment of fertility in adult animals [8]. G-protein coupled receptors, in particular, prokineticin 2 (PROK2) and its receptor (PROKR2) were shown to modulate GnRH neuron migration. The mice null for PROKR2 had a loss of GnRH neurons in the forebrain on embryonic day 13.5 (ED13.5) and in adults, had formed a huge and tangled web of olfactory vomeronasal axons that would alter GnRH neuron movement [9].
3. Development of the GnRH System in Different Pathological States
Disorders in the development of the GnRH system lead to impaired puberty and fertility in adults. The reasons for the development of reproductive system disorders in most patients are still not fully determined. Genetic mutations in genes that determine the synthesis of factors involved in the migration of GnRH neurons are detected in no more than 50% of patients [10]. Despite the fact that the proportion of certain genetic causes of underdevelopment of the reproductive system is growing, adverse factors affecting the developing human body in early ontogenesis can significantly influence the development of the GnRH system [11][12][13][14]. The best known disease associated with impaired migration of GnRH neurons is the so-called Kallmann syndrome. The diagnosis of Kallmann syndrome is based on the identification of impairments in sexual development: a decrease in the mass of the gonads, as well as low secretion of gonadotropins and sex steroids, and in addition, these symptoms are accompanied by a loss of smell (anosmia). When there is Kallmann syndrome in a human fetus with a genetic mutation in the X-linked gene KAL1, there is a complete impairment of the penetration of GnRH neurons into the brain, with neurons located in the nasal region along the dorsal surface of the ethmoid plate of the ethmoid bone [15]. Receptor, which is activated by the prokineticins (PRKR2), and the mouse nasal embryonic GnRH factor gene (NELF) are required for olfactory axonal outgrowth and GnRH neuronal migration in mice [16].
Reproductive impairment caused by decreased GnRH secretion is not always associated with impaired neuronal migration. Abnormal development of the gonads without defects in the development of the olfactory system in humans is called idiopathic hypogonadotropic hypogonadism. This pathology is more common in men and manifests itself in the early postnatal period [17]. The absence of puberty against the background of a decrease in GnRH secretion was also found in women [17].
For studies of pathological processes developing in the reproductive system, hypogonadal mice (hpg mice) are widely used. These mice have a general underdevelopment of the gonads and disorders of the reproductive system. They have a spontaneous deletion mutation of 33.5 kilobases encompassing the distal half of the GNRH1 gene, which completely disrupts the transcription and synthesis of GnRH, leading to a lifelong deficiency of gonadotropins and sex steroids. In male hpg mice, changes in the concentration of amyloid precursors, as well as a decrease in the level of IL-1β in the hippocampus and choline acetyltransferase per neuron, were revealed, similar to changes in patients with Alzheimer’s disease [18].
Currently, more and more clinical and experimental data are coming in on the negative influence of various unfavorable factors on the developing fetus, including the HPG axis. Their impact leads to persistent changes in the epigenetic regulation of gene transcription, and as a consequence, to phenotypic changes [19][20]. The HPG axis disorders are based on epigenetic modification of the estrogen receptor (ERα) gene promoter and subsequent changes in the expression of this gene [21][22]. It has been shown that estrogens increase the expression of the KISS1 gene in the brain of mice by the acetylation of histones in its promoter region in the anteroventral periventricular nucleus of the hypothalamus and suppress its expression by the deacetylation of histones in the arcuate nucleus. Epigenetic regulation of the KISS1 gene by estrogen-positive feedback induces the release of GnRH [23]. Suppression of KISS1 gene expression in female rats in the medial preoptic region of the hypothalamus, induced by systemic inflammation in the neonatal period, significantly slows down the onset of puberty [24].
One of the risk factors is a bacterial infection that induces inflammation in both the mother and the fetus. Bacterial infections, particularly asymptomatic infections during pregnancy that are not normally treated, can lead to serious complications including pre-term parturition, low birth weight, and CNS damage [25][26][27][28].
Experimental studies often use LPS, a major component of the outer membrane in bacteria. LPS is a strong inducer of innate immunity consisting primarily of cytokine induction, inflammation, fever, complement cascade activation, hypothalamic–pituitary–adrenal (HPA) axis activation, and sickness behavior [29][30]. By inducing an immune response in the mother, LPS is able to alter the level of cytokines in the fetus. Proinflammatory cytokines are probably a link between maternal infection and subsequent disruptions in the development and further functioning of various brain systems in the offspring.
According to our data, activation of the immune system of rats or mice by LPS (Escherichia coli) on the 12th day of pregnancy leads to a decrease in the rate of migration of GnRH neurons from the nasal region to the brain and is accompanied by an increase in the level of IL-6, LIF, and MCP-1 in the blood of the mother and the fetuses [14]. In sexually mature offspring, there is a decrease in the level of GnRH in the hypothalamus and a reduction in the level of gonadotropins and sex steroids in the peripheral blood. The effect of immunological stress on the development of the fetal GnRH system depends on the period of exposure. If such activation takes place at the initial stage of GnRH neuronal migration in the fetus, it results in the overall disorganization of this process. At the same time, LPS administration to pregnant females at subsequent stages of neuronal migration does not lead to a redistribution of GnRH neurons in the fetal brain, which indicates that some other mechanisms become involved in the regulation of their migration [31].
The appearance of GnRH neurons in the forebrain with a delay, apparently, causes changes in the formation of the necessary axonal connections, which leads to impairments at key points in the development of the HPG axis. In adult animals, LPS-induced inflammation has been proposed to suppress axonal transport of GnRH mRNA to GnRH neurons in the preoptic region and the median eminence of the hypothalamus, where their terminals are projected and GnRH mRNA is detected. The formation of a synaptic network that modulates the function of GnRH neurons is controlled by the integrative activity of internal and external signals. Thus, suppression of GnRH synthesis in the hypothalamus and suppression of gonadotropin secretion in the pituitary gland after systemic administration of LPS to rats are accompanied by an increase in IL-1β secretion and tumor necrosis factor (TNF) α in the medial preoptic region of the hypothalamus [32]. It is known that an increased level of TNFα in the blood induces sepsis and in the brain, it causes apoptosis of developing oligodendrocytes, mediated by the apoptosis-inducing factor (AIF), which is translocated into the nucleus under the influence of TNFα [33][34]. An LPS-induced intrauterine inflammation in mice, accompanied by increased levels of TNFα, leads to hypomyelination and diffuse brain damage, both in the white matter and in the gray matter of fetuses, including the hypothalamus, the thalamus, and the hippocampus [35].
With the central administration of IL-1β, an increase in the levels of β-endorphin and tachykinins is observed. They are involved in the retransmission of the cytokine signal into GnRH neurons and suppress their functioning [36]. IL-1α and granulocyte macrophage colony-stimulating factor (GM-CSF) block NO-induced GnRH secretion in the mediobasal region of the hypothalamus, which in turn blocks the pulsatile secretion of LH into the blood and suppresses GnRH-regulated sexual behavior [37]. Through GnRH neurons, various neurotransmitters and neuropeptides, such as monoamines, GABA, neuropeptide Y, opioids, cytokines, KISS1, as well as leptin, transmit external stimulus signals that affect the state of the HPG system [38][39][40].
An LPS-induced inflammation in the mother during pregnancy causes impairments in the formation of GABA-, dopamine- and serotonin-producing neurons in the developing fetal brain [28]. Administration of LPS to female rats on the 11th day of gestation leads to a decrease in the number of dopaminergic neurons and an increase in the activity of microglia, as well as to an increase in the level of proinflammatory cytokines, mainly TNFα, in the substantia nigra in postnatal offspring [41]. Moreover, LPS can influence the differentiation of monoaminergic neurons not only in the brain stem, but also in other brain structures, including the hypothalamus in fetuses. After the introduction of LPS, the following was observed: a decrease in the expression of the enzyme synthesizing dopamine (tyrosine hydroxylase) in the substantia nigra and of the enzyme synthesizing serotonin (tryptophan hydroxylase) in the dorsal raphe nucleus, a decrease in the levels of dopamine and serotonin in the olfactory bulbs of the frontal cortex, the nucleus accumbens, the striatum, the amygdala, the hippocampus, and the hypothalamus, as well as a decrease in the expression of the enzyme synthesizing GABA and reelin in the dentate gyrus and the CA1 in offspring [42][43].
Thus, activation of the immune system in early ontogenesis triggers a cascade of intermediators that cause impairments in the formation of both the immune system and the HPG system, which leads to an increased risk of immunological, behavioral, and reproductive disorders in offspring.
References
- Wakabayashi, Y.; Yamamura, T.; Sakamoto, K.; Mori, Y.; Okamura, H. Electrophysiological and Morphological Evidence for Synchronized GnRH Pulse Generator Activity among Kisspeptin/neurokinin B/dynorphin A (KNDy) Neurons in Goats. J. Reprod. Dev. 2013, 59, 40–48.
- Comninos, A.N.; Anastasovska, J.; Sahuri-Arisoylu, M.; Li, X.; Li, S.; Hu, M.; Jayasena, C.N.; Ghatei, M.A.; Bloom, S.R.; Matthews, P.M.; et al. Kisspeptin signaling in the amygdala modulates reproductive hormone secretion. Brain Struct. Funct. 2016, 221, 2035–2047.
- Yip, S.H.; Boehm, U.; Herbison, A.E.; and Campbell, R.E. Conditional viral tract tracing delineates the projections of the distinct kisspeptin neuron populations to gonadotropin-releasing hormone (GnRH) neurons in the mouse. Endocrinololy 2015, 156, 2582–2594.
- Lehman, M.N.; Coolen, L.M.; Goodman, R.L. Minireview: Kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: A central node in the control of gonadotropin-releasing hormone secretion. Endocrinology 2010, 151, 3479–3489.
- Moore, A.M.; Coolen, L.M.; Porter, D.T.; Goodman, R.L.; Lehman, M.N. KNDy cells revisited. Endocrinology 2018, 159, 3219–3234.
- Igaz, P.; Salvi, R.; Rey, J.P.; Glauser, M.; Pralong, F.P.; Gaillard, R.C. Effects of cytokines on gonadotropin-releasing hormone (GnRH) gene expression in primary hypothalamic neurons and in GnRH neurons immortalized conditionally. Endocrinology. 2006, 147, 1037–1043.
- Yoshida, K.; Tobet, S.A.; Crandall, J.E.; Jimenez, T.P.; Schwarting, G.A. The migration of luteinising hormone-releasing hormone neurons in the developing rat is associated with a transient, caudal projection of the vomeronasal nerve. J. Neurosci. 1995, 15, 7769–7777.
- Parkash, J.; Cimino, I.; Ferraris, N.; Casoni, F.; Wray, S.; Cappy, H.; Prevot, V.; Giacobini, P. Suppression of β1-Integrin in gonadotropin-releasing hormone cells disrupts migration and axonal extension resulting in severe reproductive alterations. J. Neurosci. 2012, 32, 16992–17002.
- Matsumoto, S.; Yamazaki, C.; Masumoto, K.H.; Nagano, M.; Naito, M.; Soga, T.; Hiyama, H.; Matsumoto, M.; Takasaki, J.; Kamohara, M.; et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc. Natl. Acad. Sci. USA 2006, 103, 4140–4145.
- Balasubramanian, R.; Crowley, W.F., Jr. Isolated GnRH deficiency: A disease model serving as a unique prism into the systems biology of the GnRH neuronal network. Mol. Cell. Endocrinol. 2011, 346, 4–12.
- Tanriverdi, F.; Silveira, L.F.G.; MacColl, G.S.; Bouloux, P.M.G. The hypothalamic-pituitary-gonadal axis: Immune function and autoimmunity. J. Endocrinol. 2003, 176, 293–304.
- Izvolskaia, M.S.; Tillet, Y.; Sharova, V.S.; Voronova, S.N.; Zakharova, L.A. Disruptions in the hypothalamic-pituitary-gonadal axis in rat offspring following prenatal maternal exposure to lipopolysaccharide. Stress 2016, 19, 198–205.
- Wu, X.Q.; Li, X.F.; Ye, B.; Popat, N.; Milligan, S.R.; Lightman, S.L.; O’Byrne, K.T. Neonatal programming by immunological challenge: Effects on ovarian function in the adult rat. Reproduction 2011, 41, 241–248.
- Sharova, V.S.; Izvol’skaia, M.S.; Voronova, S.N.; Zakharova, L.A. Effect of bacterial endotoxin on migration of gonadotropin-releasing, hormone producing neurons in rat embryogenesis. Ontogenez 2011, 42, 439–446.
- Cadman, S.M.; Kim, S.H.; Hu, Y.; González-Martínez, D.; Bouloux, P.M. Molecular pathogenesis of Kallmann’s syndrome. Horm. Res. 2007, 67, 231–242.
- Gianetti, E.; Tusset, C.; Noel, S.D.; Au, M.G.; Dwyer, A.A.; Hughes, V.A.; Crowley, W.F.; Kaiser, U.B.; Latronico, A.C.; Seminara, S.B. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J. Clin. Endocrinol. Metab. 2010, 95, 2857–2867.
- Shaw, N.D.; Seminara, S.B.; Welt, C.K.; Au, M.G.; Plummer, L.; Hughes, V.A.; Dwyer, A.A.; Martin, K.A.; Quinton, R.; Mericq, V.; et al. Expanding the phenotype and genotype of female GnRH deficiency. J. Clin. Endocrinol. Metab. 2011, 96, 566–576.
- Drummond, E.S.; Martins, R.N.; Handelsman, D.J.; Harvey, A.R. Altered expression of Alzheimer’s disease-related proteins in male hypogonadal mice. Endocrinology 2012, 153, 2789–279.
- Lillycrop, K.A.; Slater-Jefferies, J.L.; Hanson, M.A.; Godfrey, K.M.; Jackson, A.A.; Burdge, G.C. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 2007, 97, 1064–1073.
- Aiken, C.E.; Ozann, S.E. Sex differences in developmental programming models. Reproduction 2013, 145, R1–R13.
- Cameron, N.M.; Shahrokh, D.; Del Corpo, A.; Dhir, S.K.; Szyf, M.; Champagne, F.A.; Meaney, M.J. Epigenetic programming of phenotypic variations in reproductive strategies in the rat through maternal care. J. Neuroendocrinol. 2008, 20, 795–801.
- Champagne, F.A.; Curley, J.P. Maternal regulation of estrogen receptor alpha methylation. Curr. Opin. Pharmacol. 2008, 8, 735–739.
- Tomikawa, J.; Uenoyama, Y.; Ozawa, M.; Fukanuma, T.; Takase, K.; Goto, T.; Okamura, H.; Maeda, K.; Tsukamura, H. Epigenetic regulation of Kiss1 gene expression mediating estrogen-positive feedback action in the mouse brain. Proc. Natl. Acad. Sci. USA 2012, 109, E1294–E1301.
- Knox, A.M.; Li, X.F.; Kinsey-Jones, J.S.; Wilkinson, E.S.; Wu, X.Q.; Cheng, Y.S.; Milligan, S.R.; Lightman, S.L.; O’Byrne, K.T. Neonatal lipopolysaccharide exposure delays puberty and alters hypothalamic Kiss1 and Kiss1r mRNA expression in the female rat. J. Neuroendocrinol. 2009, 21, 683–692.
- Cai, Z.; Pan, Z.L.; Pang, Y.; Evans, O.B.; Rhodes, P.G. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr. Res. 2000, 47, 64–72.
- Gilstrap, L.C.; Ramin, S.M. Urinary tract infections during pregnancy. Obstet. Gynecol. Clin. N. Am. 2001, 28, 581–591.
- Hagberg, H.; Gressens, P.; Mallard, C. Inflammation during fetal and neonatal life: Implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 2012, 71, 444–457.
- Wang, H.L.; Pei, D.E.; Yang, R.D.; Wan, C.L.; Ye, Y.M.; Peng, S.S.; Zeng, Q.Q.; Yu, Y. Prenatal maternal vaginal inflammation increases anxiety and alters HPA axis signalling in adult male mice. Int. J. Dev. Neurosci. 2019, 75, 27–35.
- Izvolskaia, M.S.; Sharova, V.S.; Zakharova, L.A. Perinatal Inflammation Reprograms Neuroendocrine, Immune, and Reproductive Functions: Profile of Cytokine Biomarkers. Inflammation 2020, 43, 1175–1183.
- Izvolskaia, M.S.; Sharova, V.S.; Ignatiuk, V.M.; Voronova, S.N.; Zakharova, L.A. Abolition of prenatal lipopolysaccharide-induced reproductive disorders in rat male offspring by fulvestrant. Andrologia 2019, 51, e13204.
- Sharova, V.S.; Izvolskaia, M.S.; Zakharova, L.A. Lipopolysaccharide-induced maternal inflammation affects the gonadotropin-releasing hormone neuron development in fetal mice. Neuroimmunomodulation 2015, 22, 222–232.
- Watanobe, H.; Hayakawa, Y. Hypothalamic Interleukin-1 and Tumor Necrosis Factor, but not Interleukin-6, mediate the endotoxin-induced suppression of the reproductive axis in rats. Endocrinology 2003, 144, 4868–4875.
- Jurewicz, A.; Matysiak, M.; Tybor, K.; Kilianek, L.; Raine, C.S.; Selmaj, K. Tumour necrosis factor-induced death of adult human oligodendrocytes is mediated by apoptosis inducing factor. Brain 2005, 128, 2675–2688.
- Kothari, N.; Bogra, J.; Abbas, H.; Kohli, M.; Malik, A.; Kothari, D.; Srivastav, S.; Singh, P.K. Tumor necrosis factor gene polymorphism results in high TNF level in sepsis and septic shock. Cytokine 2013, 61, 676–681.
- Ginsberg, Y.; Khatib, N.; Weiner, Z.; Beloosesky, R. Maternal inflammation, fetal brain implications and suggested neuroprotection: A summary of 10 years of research in animal models. Rambam Maimonides Med. J. 2017, 8, e0028.
- Kalra, P.S.; Edwards, T.G.; Xu, B.; Jain, M.; Kalra, S.P. The anti-gonadotropic effects of cytokines: The role of neuropeptides. Domes. Anim. Endocrinol. 1998, 15, 321–332.
- McCann, S.M.; Kimura, M.; Karanth, S.; Yu, W.H.; Mastronardi, C.A.; Rettori, V. The mechanism of action of cytokines to control the release of hypothalamic and pituitary hormones in infection. Ann. N. Y. Acad. Sci. 2000, 917, 4–18.
- Zakharova, L.A.; Izvolskaia, M.S. Interactions between the reproductive and immune systems during ontogenesis: The role of GnRH, sex steroids and immunomediators. In Sex Steroids; Kahn, S.M., Ed.; InTech: Rijeka, Croatia; London, UK, 2012; pp. 341–370.
- Uenoyama, Y.; Inoue, N.; Kei-Maeda, I.; Tsukamura, H. The roles of kisspeptin in the mechanism underlying reproductive functions in mammals. J. Reprod. Dev. 2018, 64, 469–476.
- Herman, A.P.; Tomaszewska-Zaremba, D. Effect of endotoxin on the expression of GnRH and GnRHR genes in the hypothalamus and anterior pituitary gland of anestrous ewes. Anim. Reprod. Sci. 2010, 120, 105–111.
- Wang, S.; Yan, J.Y.; Lo, Y.K.; Carvey, P.M.; Ling, Z. Dopaminergic and serotoninergic deficiencies in young adult rats prenatally exposed to the bacterial lipopolysaccharide. Brain Res. 2009, 1265, 196–204.
- Nouel, D.; Burt, M.; Zhang, Y.; Harvey, L.; Boksa, P. Prenatal exposure to bacterial endotoxin reduces the number of GAD67- and reelin-immunoreactive neurons in the hippocampus of rat offspring. Eur. Neuropsychopharmacol. 2012, 22, 300–307.
- Harvey, L.; Boksa, P. A stereological comparison of GAD67 and reelin expression in the hippocampal stratum oriens of offspring from two mouse models of maternal inflammation during pregnancy. Neuropharmacology 2012, 62, 1767–1776.


