 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jost Ruwoldt | + 5665 word(s) | 5665 | 2020-11-30 09:28:38 | | | |
| 2 | Jost Ruwoldt | -17 word(s) | 5648 | 2021-01-01 13:26:12 | | | | |
| 3 | Catherine Yang | -2290 word(s) | 3358 | 2021-01-05 04:22:26 | | |
Video Upload Options
Lignosulfonates are biobased surfactants and specialty chemicals, which are usually produced as a byproduct during sulfite pulping of wood. They are the technical lignin that is, by far, the most commercially traded and are hence vital for replacing non‑renewable and fossil‑based chemicals. Due to their prominent use as plasticizers, dispersants, and stabilizers, the physicochemical properties of lignosulfonates play a key role in determining their end‑use and performance. Their chemical composition and structure are inherently linked to the characteristic behavior of lignosulfonates.
This entry hence outlines the fundamental chemistry of lignosulfonates, while discussing the following physicochemical properties:
· Solubility in different solvents
· Conformation and shape in aqueous solution
· Self-association and agglomeration in aqueous solution
· Precipitation
· Adsorption at surfaces and interfaces
1. Introduction
Lignosulfonates are generated as a by‑product during sulfite pulping of wood [1]. During this process, the “infinite” lignin network is broken down and sulfonate groups are introduced on the lignin. The degraded lignin is hence rendered water‑soluble and can be separated from the cellulosic fibers and material. Lignin isolated by a different process may alternatively be sulfonated post‑separation, e.g. as sulfonated or sulfomethylated Kraft, soda or hydrolysis lignin [2][3][4]. The original lignin structure is preserved to a certain degree, which endows lignosulfonates with its amphiphilic properties. In technical applications, lignosulfonates are usually found as the polyelectrolyte salt of the lignosulfonic acid. Anionic groups such as sulfonate and carboxylic groups ensure water solubility, while less polar groups, i.e. aromatic and aliphatic moieties, facilitate interactions with surfaces and interfaces. Due to their surface activity, lignosulfonates are considered surfactants.
The most common use is as a dispersant [5], which includes applications such as concrete plasticizers, drilling mud thinners, coal-slurry and dye dispersants [6]. In 1999, roughly 50% of the annually produced lignosulfonates were used as admixtures in concrete [7]. Other application areas include chelating and complexation agents, soil conditioning agents, floatation agents, dust binders, and emulsion stabilizers [5][6][8][9]. Emulsion stabilization with lignosulfonates requires high shear during emulsification, as the effect on interfacial tension is less than that of commercial surfactants [10]. Still, lignosulfonates can produce highly stable emulsions, which has yielded applications for example in agrochemical formulation [5]. Experimentally explored but not fully commercialized are applications such as corrosion and scale inhibitors, CO2 flooding and enhanced oil recovery, as well as polymer precursors and additives [11][12][13][14][15][16][17].
2. Chemical Composition
Lignosulfonates are described as randomly‑branched polyaromatic polyelectrolytes [18][19], which exhibit water‑solubility and surfactant‑like behavior [5][6][20]. Hydrophilicity is imparted by the presence of anionic sulfonate groups, but also by anionic carboxylate groups and (at high pH) phenolic hydroxyl groups [1]. The counterion is often a remnant from the pulping process, such as sodium, calcium, magnesium, or ammonia, which facilitates dissociation in aqueous solution. Apart from the dissociation equilibrium, the counterion may otherwise determine the physicochemical properties of lignosulfonates, for example by affecting the polymer conformation [21]. Some of the polar functional groups, that is, ketones, aldehydes, and methoxy groups, are not operative hydrophilic groups [22]. Aliphatic hydroxyl and ether groups can be intrinsically hydrophilic; however, their functionality is determined by the surrounding molecular structure [22][23][24]. Two examples of generic lignosulfonate structures are shown in Figure 1. It should be noted that these are simplifications of a more complicated picture. Lignosulfonates should be considered as statistical entities rather than classical chemical compounds, due to their polydisperse structure and molecular weight [25]. The molecular weight of lignosulfonates may span from less than 1000 g/mol to more than 400 000 g/mol in molecular weight [26][27]. Other technical lignin usually exhibits a lower Mw and a less broad distribution, as for example in case of soda lignin (1000 – 15 000 g/mol), Kraft lignin (1500 – 25 000 g/mol) or organosolv lignin (500 – 5000 g/mol) [28]. Lignosulfonate composition, structure, molecular weight distribution, and abundance of functional groups is dependent on aspects such as the biomass origin, sulfite pulping conditions, and post‑extraction fractionation and chemical modifications.
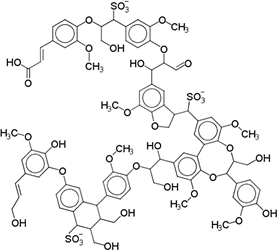
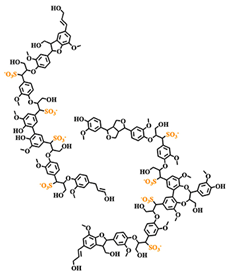
Figure 1. Generic (simplified) structure of lignosulfonates according to Kun and Pukanszky [29] (left), and Fiorani et al. [30](right).
3. Physicochemical Properties of Lignosulfonates
3.1. Solubility in Different Solvents
In contrast to other technical lignin, lignosulfonate possesses good water solubility due to an abundance of sulfonate groups [31]. Solutions of 53 wt.% in water have been reported [32], which would entail that the water‑solubility of lignosulfonate is virtually unlimited. Myrvold further studied the solubility of different lignosulfonate samples in various solvents [20][33]. The author showed that, apart from water, lignosulfonates also possess good solubility in ethylene glycol, propylene glycol, dimethyl sulfoxide (DMSO), as well as methanol‑water and dioxane-water blends with more than 20 % water. Limited solubility was reported for dimethyl formamide, methanol, cyclohexylamine, and acetic acid. It was concluded that hardwood lignosulfonates have Hansen solubility parameters further away from water than softwood lignosulfonates [20][33]. Solubility in ionic liquids at 90 °C has furthermore been demonstrated, such as choline acetate, tributylmethylphosphoniummethyl sulfate or N-butyl-N-methylpyrrolidinium dicyanamide [34].
3.2. Conformation and Shape in Aqueous Solution
An early model of the lignosulfonate conformation in aqueous solution was given by Rezanowich and Goring [35]. Based on light scattering and viscosity measurements, a microgel model was developed. The authors further proposed that the free charges were only located on the surface of the spherical molecule, as is illustrated in Figure 2. These assumptions were later refuted or refined. Following the polyelectrolyte expansion of lignosulfonates in dependence of molecular weight, Myrvold concluded that the randomly branched polyelectrolyte model provided the best description [18]. This suggested that the lignosulfonate is not a microgel structure. In addition, the spherical conformation only approximated the shape of low molecular lignosulfonate. At high molecular weight, the shape was better approximated by an “elongated or fully stretched shape” [18]. The conformation of lignosulfonates aqueous solution may indeed be better described as oblate spheroid shape[36][37].
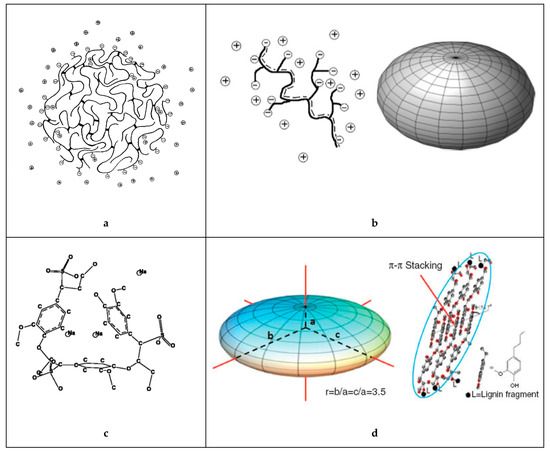
Figure 2. Idealized structure and shape of the sodium lignosulfonate macromolecule in solution according to (a) Rezanowich et al. [35] (top-left), (b) Myrvold [18]and Lauten et al. [5] (top-right), (c) Valencia et al. [21] (bottom-left) and (d) Qian et al. [37] (bottom-right).
At low salinity and low lignosulfonate concentration, the lignosulfonate molecule is expanded due to electrostatic repulsion between the anionic groups [38]. This expansion is dependent on ionic strength, as for example increasing counterion concentrations can reduce the dissociation‑associated equilibrium. In addition, charge screening occurs at high salinity. Both effects reduce electrostatic repulsion and therefore lessen the degree of expansion. An additional effect governing polyelectrolyte expansion is the pH dependent dissociation. Lignosulfonate’s sulfonate groups are mostly dissociated above pH 2, however, the carboxylic groups ionize at about pH 3-4 and the phenolic groups at around pH 9-10 [39][40]. Increasing the pH from 2 to 10 was hence reported to increase the molecular dimensions [41]. Li et al. furthermore showed in dynamic light scattering experiments that the hydrodynamic radius of lignosulfonate molecules decreased at increasing temperature [42]. The entropy is higher at elevated temperature, which enables a larger number of possible conformations, thus also reducing the average molecular dimensions [38].
3.3. Self-Association and Agglomeration in Aqueous Solution
As any system that strives to minimize the total potential energy, lignosulfonates can aggregate in aqueous solution. It has long been suggested that this aggregation is the result of hydrophobic interactions [43], where the hydrophobic moieties are oriented towards the aggregate core and the hydrophilic moieties are concentrated on the aggregate surface. As a recent study on fluorescence excitation spectra showed, sodium lignosulfonate tends to form oriented π‑π‑stacking with the spectroscopic characteristics of J‑aggregates [44]. In addition, hydrogen bonding has been suggested as a mechanism for lignosulfonate‑lignosulfonate attractive interactions [36][42]. Vainio et al. performed experiments on small‑angle X‑ray scattering, which suggested that lignosulfonate molecules aggregate on the long edges into flat aggregates [45].
Reports are diverging on the geometry of the aggregates. One report stated that lignosulfonate aggregates are nearly spherical [43], whereas another report illustrated a hollow configuration [46]. As Myrvold demonstrated, aggregation and disaggregation of lignosulfonate is highly dependent on the preparation method and parameters, such as temperature, pH, equilibration time, and concentration [47]. Both a condensed nearly‑spherical and an open‑hollow configuration are therefore realistic.
Two schematics of the proposed mechanisms during lignosulfonate aggregation are shown in Figure 3. It should be noted that the geometrical representation for individual lignosulfonate molecules used by Vainio et al. [45], that is, flat cuboid like particles, is not consistent with the spheroidal conformation reported by other authors [5][8][17].
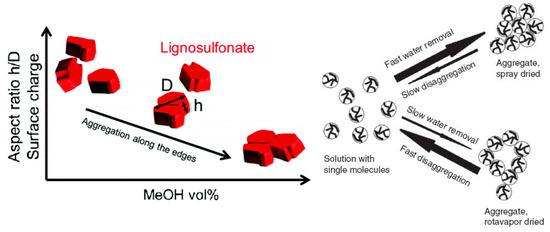
Figure 3. Lignosulfonate aggregation mechanism as proposed by Vaninio et al. [45] (left) or by Myrvold [47] (right).
Lignosulfonate aggregation can be induced by increasing lignosulfonate or salt concentration, by adding alcohol, by reducing pH, and by increasing the temperature [41][45][48]. A common denominator among most of these actions is that electrostatic repulsion is reduced, which may further enhance hydrophobic interactions.
Increasing the concentration of lignosulfonate or of another added electrolyte will increase the ionic strength. This can induce Coulomb shielding of the anionic groups or yield a lower degree of dissociation. Electrostatic repulsion between individual lignosulfonate molecules is reduced and less hydrophilic moieties will be more exposed due to a larger number of possible conformations [38]. Both effects can facilitate hydrophobic interactions, which can lead to aggregation. Adding an alcohol solvent to the aqueous solution can have a similar effect, as the overall dielectric constant is reduced leading to a lower degree of dissociation of the anionic groups. This has been shown for example by addition of methanol, which yielded a reduction of the relative permittivity in proximity of lignosulfonate molecules [45].
Lignosulfonate aggregation was also reported to be caused by a temperature increase to 38 °C and above [48]. This was accompanied by a reduction of the zeta potential [48], which would be coherent with a lower degree of dissociation [49]. As discussed previously, increasing the temperature will reduce the polyelectrolyte expansion of lignosulfonates, as a higher number of conformations become thermodynamically possible [38][42]. Such behavior would naturally facilitate aggregation, as the hydrophobic moieties become more exposed. It is interesting to note that the overall charge was affected, even though the differences in dielectric constant and dissociation constant are small [38].
The pH can especially affect the dissociation of anionic functional groups in lignosulfonates. Tang et al. reported that disaggregation of sodium lignosulfonate can occur above pH 10.34 [41]. It was argued that ionization of the phenolic groups increased electrostatic repulsion between the lignosulfonate molecules, leading to breakup of the aggregates. These results are in contrast to Yan et al., who found that increasing the pH from 3 to 12 yielded a steady increase of reduced viscosity [40]. All in all, lowering the pH could in theory promote aggregation, however, precipitation is more commonly observed as the result of pH reduction.
Classical surfactants are composed of a hydrophilic head and a lipophilic tail. This orderly arrangement accounts for rather defined properties, such as the critical micelle concentration (CMC). The analogous counterpart of lignosulfonates would be the critical aggregation concentration (CAC). Overall, reports on the CAC value of lignosulfonates are not consistent. Fluorescence spectrometry detected values between 0.15 – 0.24 g/l in one case [50] and 0.05 g/l in another case [40]. Rana et al. reported CAC values of 10 - 19 wt.% deduced from surface tension measurements [51]. Park et al. determined a CAC of 24.8 g/l by using the same technique [52]. Qiu et al. determined a CAC of 0.38 g/l via UV‑spectrometry [46]; however, the validity of this measurement is questionable, since UV‑spectrometry does not provide a linear response (absorbance increase per concentration increment) at high lignosulfonate concentrations. Overall, lignosulfonate aggregation and de‑aggregation is a kinetic process that is especially affected by the origin and composition of the sample [47]. A certain difference in CAC is therefore to be expected between authors, who performed their measurements on dissimilar samples.
3.4. Precipitation
Lignosulfonate precipitation from solution can be caused by a number of changes that destabilize its solubility. The addition of another electrolyte can invoke precipitation by salting out [10]. As has been reported, the salting out tendency for various ions is in line with both the Schulze‑Hardy rule and the Hofmeister series [10][32], with the exception of a few ions. It was further discussed that the observed effects could not be explained by the common ion effect or screening effects. Adding a solvent to an aqueous solution can cause precipitation of lignosulfonates as well [45][53], as there are many solvents that are miscible with water but pose as poor solvents for lignosulfonates [20]. Precipitation of technical lignin by pH reduction is a common method, for example to separate the lignin from alkali black liquor during wood pulping [1]. At pH 3 or lower, both the phenolic and carboxylic acid groups are mostly undissociated [39][40], which can lead to lignosulfonate precipitation if the ratio of sulfonic to carboxylic acid groups is too low.
3.5. Adsorption at Surfaces and Interfaces
Adsorption and desorption of lignosulfonates follow a similar behavior as that of other surfactants. Langmuir isotherm has been reported by several authors to describe the equilibrium adsorption of lignosulfonates on solids [54][55][56][57]. At the water‑air surface or water‑oil interface, lignosulfonate adsorption is evident by a decrease in surface or interfacial tension [10][58][59]. This decrease follows a linear‑logarithmic progression with increasing lignosulfonate concentration [10][58], but above a certain concentration the effect can decrease in slope, which has been related to the aggregation onset by some authors [51][52]. Figure 4 exhibits two comparisons of surface tension plots for lignosulfonates with other surfactants and polymers.
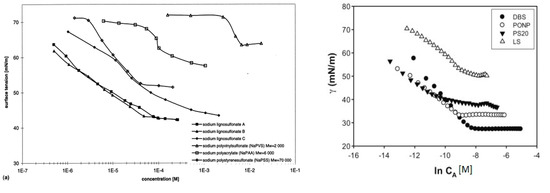
Figure 4. Equilibrium surface tension (water-air) in dependence of surfactant or polymer concentration. Comparison of lignosulfonates (LS) with polyelectrolyte polymers (left) [58] or with the surfactants dodecyl benzenesulfonate (DBS), nonylphenyl polyoxyethylene glycol (PONP, Ingepal CO-720) and polysorbate 20 (PS20, Tween 20) (right) [52].
Overall, lignosulfonate addition caused a larger reduction of surface tension than polyelectrolyte polymers, such as sodium polyacrylate or sodium polystyrenesulfonate [58]. On the other hand, less reduction of surface or interfacial tension than regular surfactants was reported [52][60][61], such as sodium dodecyl sulfate or nonylphenyl polyoxyethylene glycol. Reports are contradictory on the effect of molecular weight. One study stated that lignosulfonates with a lower average molecular weight displayed a tendency to induce larger changes of interfacial tension [10], whereas another reported that with increasing molecular weight the effect on surface tension became stronger [62]. The reduction of surface or interfacial tension can further be enhanced by increasing ionic strength or by reducing the pH [40][59][63].
Measurements of surface or interfacial tension are also instrumental for studying the kinetics of lignosulfonate adsorption. Several hours or more are usually needed to attain an equilibrium state [60][63][64]. To explain this comparably long equilibration time, it was proposed that lignosulfonate molecules undergo diffusion exchange at the interface, and that individual molecules are subject to rearrangement with respect to the interface and to each other [63]. Such conformational realignment has for example been described for petroleum asphaltenes at the water‑oil interface [65][66]. Both lignosulfonates and asphaltenes are polybranched and exhibiting a tendency for self‑association. In addition, lignosulfonates and petroleum asphaltenes have in common that emulsions stabilized by these components require overnight storage before processing [10][67], as the emulsions would otherwise be less stable. These two species have hence been compared in terms of interfacial phenomena [63].
The dynamics of lignosulfonate adsorption at the air‑water interface was studied for example by Yan and Yang [59], who showed that the adsorption kinetics are faster at low pH or at high ionic strength. The authors furthermore measured Langmuir surface compression isotherms and proposed a generalized model, which is shown in Figure 5. This model suggested a closer packing density facilitated by increased lignosulfonate concentration or higher ionic strength. This is in agreement, for example, with findings that suggest a smaller area per molecule at the interface at high salinity as compared to the low salinity condition [10][63]. In addition, an alignment of charged moieties into the aqueous phase is suggested, while the hydrophobic moieties are partly extended into the non‑aqueous phase [59].
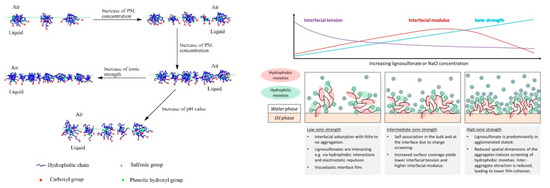
Figure 5. Model view of lignosulfonate adsorption according to Yan and Yang [59] (left) and Ruwoldt et al. [63] (right).
Fundamentally, the model view proposed by Yan and Yang is in line with the generally accepted theories; however, two statements were made that are in disagreement with other literature references, that is, the formation of a monolayer film and the adsorption of (mono-) molecular lignosulfonate were proposed [59] . Gundersen et al. also studied lignosulfonate adsorption layers using the Langmuir technique [68], based on which the formation of multilayers was proposed. Lignosulfonate multilayers were also indicated in by deposition and self‑association of lignosulfonate and cationic polymer on a solid substrate [39][69][70]. An alignment and thereby concentration of charged moieties in the aqueous phase would certainly make sense, as proposed by Yan and Yang [59], which could furthermore prevent a second or third layer from adsorbing due to electrostatic repulsion. However, lignosulfonate has proven an ability to form three dimensional aggregates also, so it would be best to assume that both mono- and multilayer formation is possible. With regards to the adsorption of (mono‑) molecular lignosulfonate, a recent study fitted the long‑time approximation of Ward and Todai to dynamic interfacial tension data [63]. The diffusion coefficients were calculated in this manner, which were several magnitudes larger than that of non‑aggregated lignosulfonate. In addition, the diffusion coefficients would decrease at increasing lignosulfonate concentration, which indicated that lignosulfonate underwent adsorption in the aggregated state. Regressions made with the Lucassen van den Tempel (LvdT) model furthermore showed a qualitatively poor fit. The shortcomings of both models suggested that some of the underlaying assumptions were insufficient, which led to the conclusion that lignosulfonate adsorption is not diffusion‑limited [63].
The kinetics of lignosulfonate adsorption on solid surfaces have been investigated by several authors [54][55][71][72][73][74]. Bai et al. studied adsorption and desorption of calcium lignosulfonate on limestone or dolomite porous media, utilizing core samples through which the surfactant solution was pumped [55][71]. A two‑step pattern was identified, which consisted of initial fast adsorption or desorption, followed by a (second) slower step. A second‑order kinetic model provided the best fit and it was concluded that desorption occurred slower than adsorption. Zulfikar et al. studied the adsorption of sodium lignosulfonate on eggshells or chitosan‑silica beads [54][72]. Pseudo second‑order kinetics fitted the data better than a first‑order kinetic model as well, and intra‑particle diffusion was reported as the rate determining step. Li et al. conducted adsorption experiments with sulfonated lignin acting as the solid (adsorbent) phase [75] A hydrogel based on sulfomethylated Kraft lignin was synthesized, onto which cationic dye (methylene blue trihydrate) was adsorbed. Interestingly, the adsorption kinetics were also of pseudo second‑order and exhibited Langmuir isotherm behavior at equilibration.
Dispersion stabilization is a frequent technical exploitation of lignosulfonate adsorption. This entails keeping a solid or liquid phase dispersed in another liquid phase, as for example in the case of concrete plasticizers (suspensions) or agrochemical formulations (emulsions) [5][76]. Several stabilization mechanisms have been proposed, which include electrostatic repulsion, stearic hindrance, particle stabilization (Pickering emulsion), and the formation of viscoelastic layers [53][63][77].
Electrostatic repulsion of droplets or particles is facilitated by the lignosulfonate adding negative charges at the surface or interface. This process can be monitored by measuring the electrophoretic mobility (zeta potential) of the dispersion, as has been done for suspensions of e.g. lead zirconate titanate [57], titanium oxide [78] or alumina [79]. Higher pH tends to induce larger changes of zeta potential, due to the higher degree of ionization of the functional groups of lignosulfonate [57][79]. It was suggested that a lower molecular mass can facilitate more efficient reduction of the zeta potential, due to screening effects of the sulfonate groups within larger lignosulfonate molecules [80]. Changes in molecular weight may, however, also be encompassed by differences in chemical make‑up and functional groups [62], which can make it difficult to delineate the effect of lignosulfonate molecular weight on zeta potential.
Experimental evidence for the formation of a viscoelastic lignosulfonate film on solids has been reported by Qin et al. using quartz crystal microbalance (QCM) [81]. Dilatational interfacial rheology and interfacial shear rheology have been used to study the viscoelastic property of lignosulfonate films at the water‑oil interface [63]. Factors such as lignosulfonate concentration, ionic strength, and type of added electrolyte were found to affect the properties of the surface or interfacial films. The film strength was studied in terms of the interfacial storage modulus and exhibited a maximum at intermediate salinity or intermediate lignosulfonate concentration. This reduction of the interfacial modulus at high salinity or high lignosulfonate concentration was attributed to either conformational changes and stearic shielding of non‑ionic groups, or bulk precipitation as induced by the addition of di‑ or trivalent cations [63]. The presence of multivalent cations increased the film strength in particular, and interfacial gelling could be induced by these.
References
- Sixta, H., Handbook of pulp. Wiley-vch: 2006.
- Konduri, M. K. R.; Fatehi, P., Production of Water-Soluble Hardwood Kraft Lignin via Sulfomethylation Using Formaldehyde and Sodium Sulfite. ACS Sustainable Chemistry & Engineering 2015, 3 (6), 1172-1182. doi: 10.1021/acssuschemeng.5b00098
- Ouyang, X.; Ke, L.; Qiu, X.; Guo, Y.; Pang, Y., Sulfonation of Alkali Lignin and Its Potential Use in Dispersant for Cement. Journal of Dispersion Science and Technology 2009, 30 (1), 1-6. doi: 10.1080/01932690802473560
- Huang, C.; Ma, J.; Zhang, W.; Huang, G.; Yong, Q., Preparation of lignosulfonates from biorefinery lignins by sulfomethylation and their application as a water reducer for concrete. Polymers 2018, 10 (8), 841. doi:
- Lauten, R. A.; Myrvold, B. O.; Gundersen, S. A., New developments in the commercial utilization of lignosulfonates. Surfactants from Renewable Resources 2010, 269-283. doi:
- Xu, C.; Ferdosian, F., Utilization of Lignosulfonate as Dispersants or Surfactants. In Conversion of Lignin into Bio-Based Chemicals and Materials, Xu, C.; Ferdosian, F., Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 2017; pp 81-90.
- Garguiak, J. D.; Lebo, S. E., Commercial Use of Lignin-Based Materials. In Lignin: Historical, Biological, and Materials Perspectives, American Chemical Society: 1999; Vol. 742, pp 304-320.
- Miretzky, P.; Cirelli, A. F., Cr(VI) and Cr(III) removal from aqueous solution by raw and modified lignocellulosic materials: A review. Journal of Hazardous Materials 2010, 180 (1), 1-19. doi: https://doi.org/10.1016/j.jhazmat.2010.04.060
- Alazigha, D. P.; Indraratna, B.; Vinod, J. S.; Heitor, A., Mechanisms of stabilization of expansive soil with lignosulfonate admixture. Transportation Geotechnics 2018, 14, 81-92. doi: https://doi.org/10.1016/j.trgeo.2017.11.001
- Ruwoldt, J.; Planque, J.; Øye, G., Lignosulfonate Salt Tolerance and the Effect on Emulsion Stability. ACS Omega 2020, 5 (25), 15007-15015. doi: 10.1021/acsomega.0c00616
- Ouyang, X.; Qiu, X.; Lou, H.; Yang, D., Corrosion and Scale Inhibition Properties of Sodium Lignosulfonate and Its Potential Application in Recirculating Cooling Water System. Industrial & Engineering Chemistry Research 2006, 45 (16), 5716-5721. doi: 10.1021/ie0513189
- Blanck, G.; Cuisinier, O.; Masrouri, F., Soil treatment with organic non-traditional additives for the improvement of earthworks. Acta Geotechnica 2014, 9 (6), 1111-1122. doi: 10.1007/s11440-013-0251-6
- Tsau, J.-S.; Syahputra, A. E.; Yaghoobi, H.; Grigg, R. B., Use of Sacrificial Agents in CO2 Foam Flooding Application. In SPE Annual Technical Conference and Exhibition, Society of Petroleum Engineers: Houston, Texas, 1999; p 9.
- Chiwetelu, C., Improving The Oil Recovery Efficacy of Lignosulfonate Solutions. PETSOC-80-03-05 1980, 19 (03), 10. doi: 10.2118/80-03-05
- Vebi, M.; Hassan, A.; Hubert, H.; Markus, B.; Ireen, G.; Robert, B.; Karin, F.; Antje, P.; Thomas, R., Lignosulfonate-based polyurethane materials via cyclic carbonates: preparation and characterization. Holzforschung 2020, 74 (2), 203-211. doi: https://doi.org/10.1515/hf-2018-0298
- Hirose, S., Novel Epoxy Resins with Unsaturated Ester Chains Derived from Sodium Lignosulfonate. Macromolecular Symposia 2015, 353 (1), 31-38. doi: 10.1002/masy.201550304
- Hu, J.-p.; Guo, M.-h., Influence of ammonium lignosulfonate on the mechanical and dimensional properties of wood fiber biocomposites reinforced with Polylactic Acid. Industrial Crops and Products 2015, 78, 48-57. doi: https://doi.org/10.1016/j.indcrop.2015.09.075
- Myrvold, B. O., A new model for the structure of lignosulphonates: Part 1. Behaviour in dilute solutions. Industrial Crops and Products 2008, 27 (2), 214-219. doi: https://doi.org/10.1016/j.indcrop.2007.07.010
- Macfarlane, A. L.; Mai, M.; Kadla, J. F., 20 - Bio-based chemicals from biorefining: Lignin conversion and utilisation. In Advances in Biorefineries, Waldron, K., Ed. Woodhead Publishing: 2014; pp 659-692.
- Myrvold, B. O., The Hansen solubility parameters of some lignosulfonates. World Acad. Sci. Eng. Technol. Trans. Energy Power Eng 2014, 1, 261. doi:
- Salazar Valencia, P. J.; Bolívar Marinez, L. E.; Pérez Merchancano, S. T., Molecular Modeling of Ammonium, Calcium, Sulfur, and Sodium Lignosulphonates in Acid and Basic Aqueous Environments. Brazilian Journal of Physics 2015, 45 (6), 567-574. doi: 10.1007/s13538-015-0364-5
- Laughlin, R. G., HLB, from a thermodynamic perspective. J Soc Cosmet Chem 1981, 32, 371-392. doi:
- Ensing, B.; Tiwari, A.; Tros, M.; Hunger, J.; Domingos, S. R.; Pérez, C.; Smits, G.; Bonn, M.; Bonn, D.; Woutersen, S., On the origin of the extremely different solubilities of polyethers in water. Nature Communications 2019, 10 (1), 2893. doi: 10.1038/s41467-019-10783-z
- Holmberg, K., Natural surfactants. Current Opinion in Colloid & Interface Science 2001, 6 (2), 148-159. doi: https://doi.org/10.1016/S1359-0294(01)00074-7
- Brauns, F. E.; Brauns, D. A., chemistry of Lignin. Supplement volume. 1960. doi:
- Fredheim, G. E.; Christensen, B. E.; Braaten, S. M., Comparison of Molecular Weight and Molecular Weight Distributions of Softwood and Hardwood Lignosulfonates. Journal of Wood Chemistry and Technology 2003, 23 (2), 197-215. doi: 10.1081/WCT-120021925
- Fredheim, G. E.; Braaten, S. M.; Christensen, B. E., Molecular weight determination of lignosulfonates by size-exclusion chromatography and multi-angle laser light scattering. Journal of Chromatography A 2002, 942 (1), 191-199. doi: https://doi.org/10.1016/S0021-9673(01)01377-2
- Vishtal, A. G.; Kraslawski, A., Challenges in industrial applications of technical lignins. BioResources 2011, 6 (3), 3547-3568. doi:
- Kun, D.; Pukánszky, B., Polymer/Lignin blends: Interactions, properties, applications. European Polymer Journal 2017, 93, 618-641. doi: https://doi.org/10.1016/j.eurpolymj.2017.04.035
- Fiorani, G.; Crestini, C.; Selva, M.; Perosa, A., Advancements and Complexities in the Conversion of Lignocellulose Into Chemicals and Materials. Frontiers in Chemistry 2020, 8, 797. doi:
- Rojas, O.; Salager, J.-L., Surface activity of bagasse Lignin derivatives found in the spent liquor of soda pulping plants. Tappi journal 1994, 77 (3), 169-174. doi:
- Myrvold, B. O., Salting-out and salting-in experiments with lignosulfonates (LSs). Holzforschung 2013, 67 (5), 549-557. doi:
- Myrvold, B. O., Differences in solubility parameters and susceptibility to salting-out between softwood and hardwood lignosulfonates. Holzforschung 2016, 70 (11), 1015-1021. doi:
- Glas, D.; Van Doorslaer, C.; Depuydt, D.; Liebner, F.; Rosenau, T.; Binnemans, K.; De Vos, D. E., Lignin solubility in non-imidazolium ionic liquids. Journal of Chemical Technology & Biotechnology 2015, 90 (10), 1821-1826. doi: 10.1002/jctb.4492
- Rezanowich, A.; Goring, D. A. I., Polyelectrolyte expansion of a Lignin sulfonate microgel. Journal of Colloid Science 1960, 15 (5), 452-471. doi: https://doi.org/10.1016/0095-8522(60)90049-0
- Vainio, U.; Lauten, R. A.; Serimaa, R., Small-Angle X-ray Scattering and Rheological Characterization of Aqueous Lignosulfonate Solutions. Langmuir 2008, 24 (15), 7735-7743. doi: 10.1021/la800479k
- Qian, Y.; Deng, Y.; Guo, Y.; Li, H.; Qiu, X., Light scattering characterization of lignosulfonate structure in saline solutions. Holzforschung 2015, 69 (4), 377-383. doi:
- Myrvold, B. O., The Polyelectrolyte behavior of randomly branched lignosulfonates. Tappi Journal 2007, 6 (11), 10-14. doi:
- Deng, Y.; Wu, Y.; Qian, Y.; Ouyang, X.; Yang, D.; Qiu, X., Adsorption and desorption behaviors of lignosulfonate during the self-assembly of multilayers. BioResources 2010, 5 (2), 1178-1196. doi:
- Yan, M.; Yang, D.; Deng, Y.; Chen, P.; Zhou, H.; Qiu, X., Influence of pH on the behavior of lignosulfonate macromolecules in aqueous solution. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2010, 371 (1), 50-58. doi: https://doi.org/10.1016/j.colsurfa.2010.08.062
- Tang, Q.; Zhou, M.; Yang, D.; Qiu, X., Effects of pH on aggregation behavior of sodium lignosulfonate (NaLS) in concentrated solutions. Journal of Polymer Research 2015, 22 (4), 50. doi: 10.1007/s10965-015-0689-3
- Li, H.; Deng, Y.; Ye, H.; Xiao, L.; Qiu, X., Effect of temperature on Polyelectrolyte expansion of lignosulfonate. BioResources 2015, 10 (1), 575-587. doi:
- Rezanowich, A.; Yean, W. Q.; Goring, D. A. I., High resolution electron microscopy of sodium Lignin sulfonate. Journal of Applied Polymer Science 1964, 8 (4), 1801-1812. doi: 10.1002/app.1964.070080429
- Deng, Y.; Feng, X.; Yang, D.; Yi, C.; Qiu, X., Pi-Pi STACKING OF THE AROMATIC GROUPS IN LIGNOSULFONATES. 2012 2012, 7 (1), 12. doi:
- Vainio, U.; Lauten, R. A.; Haas, S.; Svedström, K.; Veiga, L. S. I.; Hoell, A.; Serimaa, R., Distribution of Counterions around Lignosulfonate Macromolecules in Different Polar Solvent Mixtures. Langmuir 2012, 28 (5), 2465-2475. doi: 10.1021/la204479d
- Qiu, X.; Kong, Q.; Zhou, M.; Yang, D., Aggregation Behavior of Sodium Lignosulfonate in Water Solution. The Journal of Physical Chemistry B 2010, 114 (48), 15857-15861. doi: 10.1021/jp107036m
- Myrvold Bernt, O., Evidence for a very slow disaggregation of lignosulfonates. In Holzforschung, 2015; Vol. 69, p 9.
- Qian, Y.; Deng, Y.; Qiu, X.; Huang, J.; Yang, D., Aggregation of sodium lignosulfonate above a critical temperature. Holzforschung 2014, 68 (6), 641-647. doi:
- Kontturi, A.-K., Diffusion coefficients and effective charge numbers of lignosulphonate. Influence of temperature. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases 1988, 84 (11), 4043-4047. doi: 10.1039/F19888404043
- Li, B.; Ouyang, X. P. In Structure and properties of Lignosulfonate with different molecular weight isolated by gel column chromatography, Advanced Materials Research, Trans Tech. Publ.: 2012; pp 2024-2030.
- Rana, D.; Neale, G.; Hornof, V., Surface tension of mixed surfactant systems: lignosulfonate and sodium dodecyl sulfate. Colloid and Polymer Science 2002, 280 (8), 775-778. doi: 10.1007/s00396-002-0687-y
- Park, S.; Lee, E. S.; Sulaiman, W. R. W., Adsorption behaviors of surfactants for chemical flooding in enhanced oil recovery. Journal of Industrial and Engineering Chemistry 2015, 21, 1239-1245. doi: https://doi.org/10.1016/j.jiec.2014.05.040
- Askvik, K. M. Complexation of lignosulfonates with multivalent cations and cationic surfactants, and the impact on emulsion stability. Ph. D. Thesis, University of Bergen, Bergen, 2000.
- Zulfikar, M. A.; Wahyuningrum, D.; Lestari, S., Adsorption of Lignosulfonate Compound from Aqueous Solution onto Chitosan-Silica Beads. Separation Science and Technology 2013, 48 (9), 1391-1401. doi: 10.1080/01496395.2012.728275
- Bai, B.; Grigg, R. B., Kinetics and Equilibria of Calcium Lignosulfonate Adsorption and Desorption onto Limestone. In SPE International Symposium on Oilfield Chemistry, Society of Petroleum Engineers: The Woodlands, Texas, 2005; p 11.
- Pang, Y.-X.; Qiu, X.-Q.; Yang, D.-J.; Lou, H.-M., Influence of oxidation, hydroxymethylation and sulfomethylation on the physicochemical properties of calcium lignosulfonate. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2008, 312 (2), 154-159. doi: https://doi.org/10.1016/j.colsurfa.2007.06.044
- Ratinac, K. R.; Standard, O. C.; Bryant, P. J., Lignosulfonate adsorption on and stabilization of lead zirconate titanate in aqueous suspension. Journal of Colloid and Interface Science 2004, 273 (2), 442-454. doi: https://doi.org/10.1016/j.jcis.2004.02.044
- Askvik, K. M.; Are Gundersen, S.; Sjöblom, J.; Merta, J.; Stenius, P., Complexation between lignosulfonates and cationic surfactants and its influence on emulsion and foam stability. Colloids and Surfaces A: Physicochemical and Engineering Aspects 1999, 159 (1), 89-101. doi: https://doi.org/10.1016/S0927-7757(99)00165-X
- Yan, M.; Yang, D., Adsorption mechanism of lignosulfonate at the air/liquid interface. Journal of the Brazilian Chemical Society 2015, 26 (3), 555-561. doi:
- Simon, S.; Saadat, M.; Ruwoldt, J.; Dudek, M.; Ellis, R. J.; Øye, G., Lignosulfonates in Crude Oil Processing: Interactions with Asphaltenes at the Oil/Water Interface and Screening of Potential Applications. ACS Omega 2020, 5 (46), 30189-30200. doi: doi.org/10.1021/acsomega.0c04654
- Zaki, N. N.; Ahmed, N. S.; Nassar, A. M., SODIUM Lignin SULFONATE TO STABILIZE HEAVY CRUDE OIL-IN-WATER EMULSIONS FOR PIPELINE TRANSPORTATION. Petroleum Science and Technology 2000, 18 (9-10), 1175-1193. doi: 10.1080/10916460008949898
- Yang, D.; Qiu, X.; Pang, Y.; Zhou, M., Physicochemical Properties of Calcium Lignosulfonate with Different Molecular Weights as Dispersant in Aqueous Suspension. Journal of Dispersion Science and Technology 2008, 29 (9), 1296-1303. doi: 10.1080/01932690701866534
- Ruwoldt, J.; Simon, S.; Øye, G., Viscoelastic properties of interfacial lignosulfonate films and the effect of added electrolytes. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2020, 606, 125478. doi: https://doi.org/10.1016/j.colsurfa.2020.125478
- Ruwoldt, J.; Øye, G., Effect of Low-Molecular-Weight Alcohols on Emulsion Stabilization with Lignosulfonates. ACS Omega 2020, 5 (46), 30168-30175. doi: doi.org/10.1021/acsomega.0c04650
- Verruto, V. J.; Le, R. K.; Kilpatrick, P. K., Adsorption and Molecular Rearrangement of Amphoteric Species at Oil−Water Interfaces. The Journal of Physical Chemistry B 2009, 113 (42), 13788-13799. doi: 10.1021/jp902923j
- Freer, E. M.; Radke, C. J., RELAXATION OF ASPHALTENES AT THE TOLUENE/WATER INTERFACE: DIFFUSION EXCHANGE AND SURFACE REARRANGEMENT. The Journal of Adhesion 2004, 80 (6), 481-496. doi: 10.1080/00218460490477143
- Keleşoğlu, S.; Barrabino Ponce, A.; Humborstad Sørland, G.; Simon, S.; Paso, K.; Sjöblom, J., Rheological properties of highly concentrated dense packed layer emulsions (w/o) stabilized by asphaltene. Journal of Petroleum Science and Engineering 2015, 126, 1-10. doi: https://doi.org/10.1016/j.petrol.2014.11.031
- Gundersen, S. A.; Ese, M.-H.; Sjöblom, J., Langmuir surface and interface films of lignosulfonates and Kraft lignins in the presence of electrolyte and asphaltenes: correlation to emulsion stability. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2001, 182 (1), 199-218. doi: https://doi.org/10.1016/S0927-7757(00)00739-1
- Deng, Y.; Zhang, W.; Wu, Y.; Yu, H.; Qiu, X., Effect of Molecular Weight on the Adsorption Characteristics of Lignosulfonates. The Journal of Physical Chemistry B 2011, 115 (49), 14866-14873. doi: 10.1021/jp208312a
- Ouyang, X.; Deng, Y.; Qian, Y.; Zhang, P.; Qiu, X., Adsorption Characteristics of Lignosulfonates in Salt-Free and Salt-Added Aqueous Solutions. Biomacromolecules 2011, 12 (9), 3313-3320. doi: 10.1021/bm200808p
- Bai, B.; Wu, Y.; Grigg, R. B., Adsorption and Desorption Kinetics and Equilibrium of Calcium Lignosulfonate on Dolomite Porous Media. The Journal of Physical Chemistry C 2009, 113 (31), 13772-13779. doi: 10.1021/jp9028326
- Zulfikar, M. A.; Setiyanto, H.; Djajanti, S. D., Effect of temperature and kinetic modelling of lignosulfonate adsorption onto powdered eggshell in batch systems. Songklanakarin J. Sci. Technol 2013, 35, 1-31. doi:
- Klapiszewski, Ł.; Zdarta, J.; Szatkowski, T.; Wysokowski, M.; Nowacka, M.; Szwarc-Rzepka, K.; Bartczak, P.; Siwińska-Stefańska, K.; Ehrlich, H.; Jesionowski, T., Silica/lignosulfonate hybrid materials: Preparation and characterization. Central European Journal of Chemistry 2014, 12 (6), 719-735. doi: 10.2478/s11532-014-0523-5
- Li, H.-q.; Huang, G.-h.; An, C.-j.; Zhang, W.-x., Kinetic and equilibrium studies on the adsorption of calcium lignosulfonate from aqueous solution by coal Fly Ash. Chemical Engineering Journal 2012, 200-202, 275-282. doi: https://doi.org/10.1016/j.cej.2012.06.051
- Li, J.; Li, H.; Yuan, Z.; Fang, J.; Chang, L.; Zhang, H.; Li, C., Role of sulfonation in Lignin-based material for adsorption removal of cationic dyes. International Journal of Biological Macromolecules 2019, 135, 1171-1181. doi: https://doi.org/10.1016/j.ijbiomac.2019.06.024
- Banfill, P. F. G.; Bowen, P.; Flatt, R.; Galmiche, L.; Houst, Y.; Kauppi, A.; Lafuma, F.; Livesey, P.; Mäder, U.; Myrvold, B. In Improved superplasticisers for high performance concrete: the SUPERPLAST project, Procedings of the 12th International Congress on the Chemistry of Cement, Conseil national de recherches du Canada: 2007; p fin00344. pdf.
- Gundersen, S. A. Lignosulfonates and Kraft lignins as oil-in-water emulsion stabilizers. Department of Chemistry, University of Bergen, Bergen, 2000.
- Qiu, X.; Yan, M.; Yang, D.; Pang, Y.; Deng, Y., Effect of straight-chain alcohols on the physicochemical properties of calcium lignosulfonate. Journal of Colloid and Interface Science 2009, 338 (1), 151-155. doi: https://doi.org/10.1016/j.jcis.2009.05.072
- Megiatto, J. D.; Cerrutti, B. M.; Frollini, E., Sodium lignosulfonate as a renewable stabilizing agent for aqueous alumina suspensions. International Journal of Biological Macromolecules 2016, 82, 927-932. doi: https://doi.org/10.1016/j.ijbiomac.2015.11.004
- Ge, Y.; Li, Z.; Pang, Y.; Qiu, X., Influence of molecular mass of lignosulfonates on the resulting surface charges of solid particles. International Journal of Biological Macromolecules 2013, 52, 300-304. doi: https://doi.org/10.1016/j.ijbiomac.2012.10.018
- Qin, Y.; Qiu, X.; Liang, W.; Yang, D., Investigation of Adsorption Characteristics of Sodium Lignosulfonate on the Surface of Disperse Dye Using a Quartz Crystal Microbalance with Dissipation. Industrial & Engineering Chemistry Research 2015, 54 (49), 12313-12319. doi: 10.1021/acs.iecr.5b03582



