 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rocío Seoane | + 1014 word(s) | 1014 | 2020-12-16 08:32:08 | | | |
| 2 | Vicky Zhou | Meta information modification | 1014 | 2020-12-29 10:42:47 | | |
Video Upload Options
Cellular senescence is considered a stress response that protects cells against malignant transformation, facilitates tissue repair and development, and prevents virus replication. However, excessive accumulation of senescent cells is associated with chronic diseases such as age-related disorders, cancer, inflammatory diseases and virus replication. The relationship between virus and cellular senescence is proving to be very complex. Cellular senescence can be induced in response to virus infection restricting virus propagation. Some viruses are able to exploit the senescence program to improve their replication, while others have developed strategies to subvert senescence. Therapeutic approaches to eliminate senescent cells may be used as a mechanism to ameliorate age-related diseases, but they may have an impact on virus replication.
1. Introduction
Viral infection triggers the activation of many cellular stress–response pathways, such as heat shock, oxidative-stress or DNA damage response (DDR), that can result in the induction of apoptosis or autophagy. In most cases, these cellular responses contribute to controlling virus replication. However, some viruses have developed strategies to avoid these antiviral responses or subvert them for their own benefit [1][2]. Another consequence of the activation of stress response pathways by viruses may be the induction of senescence. For some viruses, the unspecific induction of senescence is part of the antiviral response, limiting virus replication. Reinforcing the hypothesis that senescence contributes to control virus replication, some viruses encode for proteins that specifically inhibit this process. In contrast, some viruses are able to usurp the senescence pathway to promote virus production. For example, some of the transcriptionally upregulated genes in senescent cells are cellular proteins that work as viral receptors. Moreover, the characteristic stable cell cycle arrest that defines cell senescence can benefit some viral agents. For example, the human immunodeficiency virus (HIV) is more transcriptionally active in G2 [3], and a G2 cell cycle arrest increases the number of integrated HIV provirus [4]. In addition, a G2 arrest can also modulate viral or cellular genes that are important for the completion of the virus life cycle, as proposed for the enhanced expression of papillomavirus capsid proteins of human papillomavirus type 6 (HPV6) and bovine papillomavirus type 1 (BPV1) [5]. Similar to what occurs with other antiviral responses, the outcome of senescence depends on the virus.
Cellular senescence denotes a condition of stable cell cycle arrest in which senescent cells do not replicate but stay viable and metabolically active. This process can be beneficial and protect against cancer or other types of stress, but an excess of senescent cells can favor cancer progression [6]. Senescent cells are characterized by metabolic and morphological alterations, reorganization of the chromatin, altered gene expression and the secretion of a variety of cytokines, growth factors, proteases, and chemokines called senescent-associated secretory phenotype (SASP) [7][8].
Although many biomarkers of senescence have been identified, most of them lack specificity or are restricted to particular conditions. Among the diverse hallmarks of cellular senescence, some of the most characteristic ones are a persistent DDR; a stable cell cycle arrest mediated by the p53/p21Cip1 and p16INK4a/pRb pathways; morphological changes characterized by enlarged size, flattened shape, extensive vacuolization and multinucleation; upregulation of the cell cycle regulators p16INK4a and p21Cip1; formation of senescence-associated heterochromatin foci (SAHF); the acquisition of a senescence-associated secretory phenotype (SASP); and increased levels of beta-galactosidase (SA-beta-gal) activity at pH 6.0 [9].
Cellular senescence can occur in response to different stimuli, including chemotherapeutic treatment, oxidative or genotoxic stress, ionizing radiation, telomere shortening, oncogenic signaling or virus infection [10]. DNA damage is a key event for the induction of cell senescence in response to many of these stimuli, such as treatment with chemotherapeutic drugs, oxidative stress, the shortening of telomeres, or oncogenic stress [11].
A factor linked to both DNA damage and senescence is interferon (IFN) signaling. Persistent beta IFN treatment triggers a DNA damage signaling pathway and senescence [12], and DNA damage induces type I IFN and stimulate IFN signaling, which amplifies DNA damage responses and promotes a p53-dependent senescence program [13][14][15].
2. The Interaction of Viruses with the Cellular Senescence Response
Prolonged IFN treatment or infection with some viruses can promote cellular senescence, which can protect against the infection with these or other viral agents, but it may contribute to the physiopathology of the infection. Some viral agents have developed strategies to prevent cellular senescence, thereby promoting virus replication and virus-related diseases. Others have evolved different mechanisms to exploit the senescence program for their own benefit. Senescence-targeted therapies have proved effective in removing senescent cells from animal models and humans and have been proposed as a therapeutic strategy to delay, prevent or treat different age-associated pathologies. Senolytic strategies may also be useful to combat the infection with those viruses that benefit from cellular senescence (Figure 1). One potential problem of using oncolytic viruses as senolytic agents is the antiviral immunity that can decrease the effectiveness of the viral agent. In addition, the proinflammatory response promoted by the virus will be added to the extracellular factors that comprise the SASP and that evoke immune responses. Could this strategy result in excessive inflammation? Finally, it is also important to consider that many clinical data reveal the reactivation of latent viral infections in response to senolytic drugs. What would be the effect of a virus coinfection? More studies are needed to define the benefits and risks of these compounds on virus-related diseases.
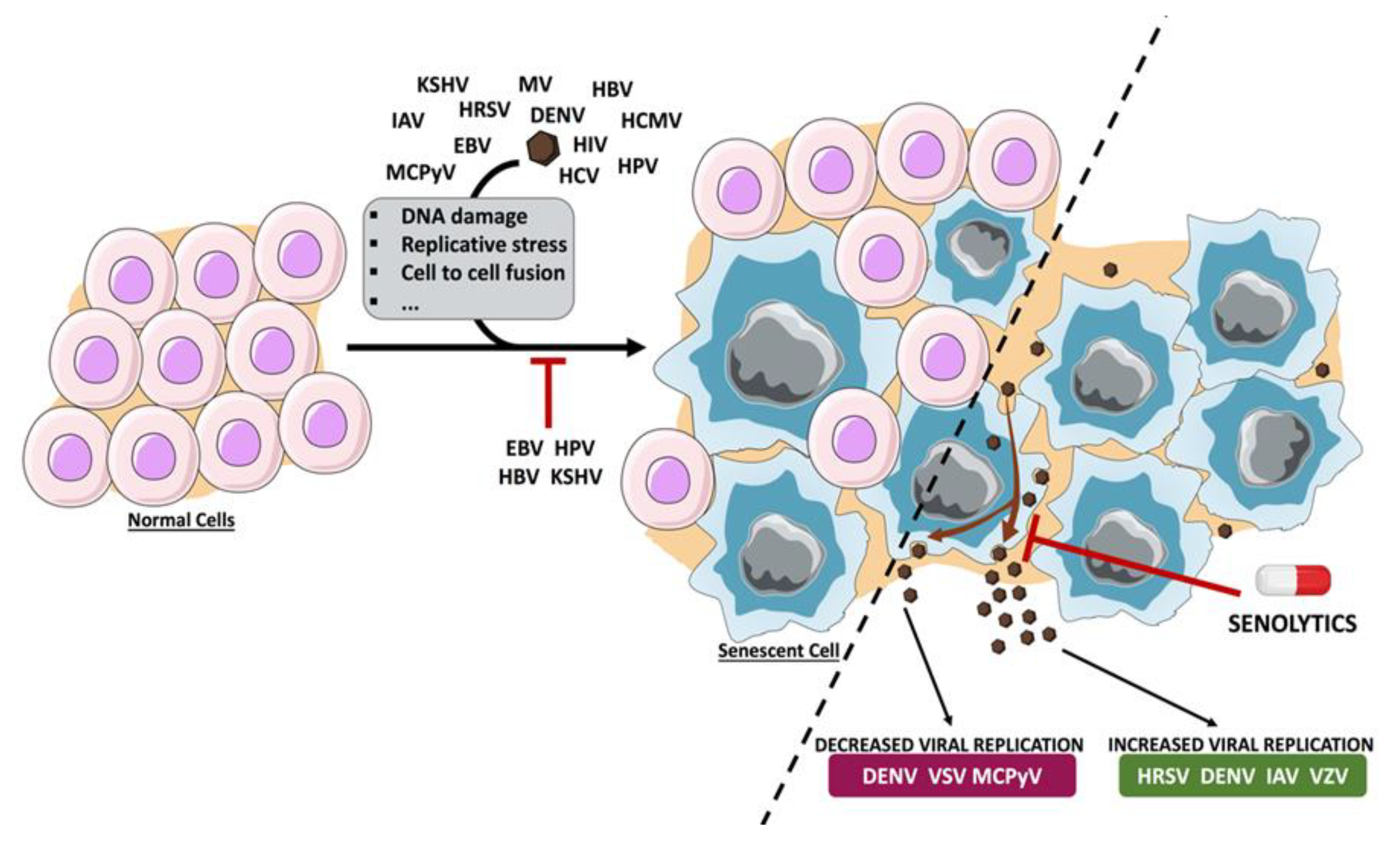
References
- Gillet, G. Viral inhibition of apoptosis. Trends Microbiol. 1996, 4, 312–317.
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Genet. 2018, 16, 341–354.
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71.
- Groschel, B.; Bushman, F.D. Cell cycle arrest in G2/M promotes early steps of infection by human immunodeficiency virus. J. Virol. 2005, 79, 5695–5704.
- Davy, C.; Doorbar, J. G2/M cell cycle arrest in the life cycle of viruses. Virology 2007, 368, 219–226.
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453.
- Hernandez-Segura, A.; Nehme, J.; DeMaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453.
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408.
- Gorgoulis, V.G.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827.
- He, S.; Sharpless, N.E. Senescence in health and disease. Cell 2017, 169, 1000–1011.
- Chen, J.-H.; Halesy, C.N.; Ozanne, S.E. DNA damage, cellular senescence and organismal ageing: Causal or correlative? Nucleic Acids Res. 2007, 35, 7417–7428.
- Moiseeva, O.; Mallette, F.A.; Mukhopadhyay, U.K.; Moores, A.; Ferbeyre, G. DNA damage signaling and p53-dependent senescence after prolonged β-interferon stimulation. Mol. Biol. Cell 2006, 17, 1583–1592.
- Brzostek-Racine, S.; Gordon, C.; Van Scoy, S.; Reich, N.C. The DNA damage response induces IFN. J. Immunol. 2011, 187, 5336–5345.
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, J.A.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343.
- Yu, Q.; Katlinskaya, Y.V.; Carbone, C.J.; Zhao, B.; Katlinski, K.V.; Zheng, H.; Guha, M.; Li, N.; Chen, Q.; Yang, T.; et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015, 11, 785–797.


