 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ting Fen Tsai | + 4152 word(s) | 4152 | 2020-12-18 02:43:36 | | | |
| 2 | Camila Xu | Meta information modification | 4152 | 2020-12-29 10:58:01 | | |
Video Upload Options
CISD2 is an evolutionally conserved protein that is mainly located in the mitochondria, ER, and MAMs. The expression level of Cisd2 decreases during natural ageing in many tissues and organs, including the heart, skeletal muscles, liver, brain, and skin.
1. Introduction
Despite the fact that the clinical management of cardiac diseases has improved over the past decades, cardiac disease remains the leading cause of mortality in an ageing population [1]. In particular, as the number of older individuals in populations has increased in many countries, a steep increase in age-related heart failure has begun to represent one of the greatest challenges confronting global health care [2]. In the United States of America, the estimated number of adults that are 20 years of age or older and have suffered from heart failure increased from 5.7 million between 2009 and 2012 to 6.2 million between 2013 and 2016 [1]; this suggests that there is similar higher prevalence all over the world [3]. The limited endogenous capacity of a heart to undergo repair/regeneration results in an accumulating burden of prior insults, which when combined with the structural and functional changes that occur during cardiac ageing, results in diminished cardiac reserves and an elevated risk of heart failure in older populations [4]. The grave prognosis for aged patients who have suffered heart failure is the result of a number of unknown molecular mechanisms that underlie the pathophysiology of cardiac ageing. However, accumulating evidence has shown that mitochondria play a central role in cardiac ageing and age-related cardiac diseases. Mitochondrial dysfunction brings about dysregulated Ca2+ homeostasis [5][6], impaired mitophagy [7][8], and metabolic inflexibility [9][10][11]. Healthy mitochondria are essential for cardiomyocytes to maintain a normal functionality in the heart.
Mitochondrial dysfunction is one of the hallmarks of ageing [12]. To meet the high-energy requirement of the heart, mitochondria occupy about 40% of the volume of cardiomyocytes [13]. Ageing affects the functioning of both mitochondria and the endoplasmic reticulum (ER), as well as having an impact on their contact sites in the cardiomyocytes, namely the mitochondria-associated membranes (MAMs). In the past decade, the markedly better understanding of the structures, tethers, and functions of MAMs has revealed the important roles that these structures play in cell physiology and ageing [14]. Four major functions of the membrane contact sites at MAMs have been identified, namely signaling, the regulation of organelle membrane dynamics, lipid transport, and metabolic channeling [14][15][16]. In an aged heart, age-related mitochondrial alterations include reduced mitochondrial Ca2+ uptake and diminished buffering capacity against reactive oxygen species (ROS); these result in impaired metabolism, which in turn bring about an increased sensitivity of the heart to stress, as well as compromising cardiac functioning. Therefore, a deeper understanding of mitochondrial and MAM functioning during cardiac ageing is pivotal to ultimately develop novel therapeutic strategies to reduce the burden of ageing.
Previously our studies have revealed that Cisd2 knockout results in a premature aging phenotype with a shorten lifespan. Furthermore, Cisd2 deficiency leads to mitochondrial dysfunction, a disruption of cytosolic Ca2+ homeostasis, elevated ROS production, and dysregulated autophagy [17][18][19][20][21][22][23]. Conversely, a persistently high level of Cisd2, which can be achieved by transgenic overexpression, is able to reverse age-related cardiac dysfunction [19], Alzheimer’s-related neuronal loss [23], nonalcoholic fatty liver disease [18], and sarcopenia [22]. It does the above by maintaining Ca2+ homeostasis, reducing ROS production, and improving metabolic flexibility. Recent genetic evidence highlights Cisd2 as a promising new target for developing novel therapeutics that are aimed at attenuating cardiac ageing and providing a new avenue to geroprotection.
2. Defective Mitochondria–Lysosomal Crosstalk during Cardiac Ageing
Cardiovascular disease is the leading cause of mortality among older adults [24]. At the structural level, age-related mitochondrial damage, which includes enlarged organelles, matrix derangement, and loss of cristae of mitochondrial inner membrane, is often found in aged hearts. At the functional level, age-related functional impairment, which includes decreased ATP production, increased ROS generation, and defective quality control, is also evident in aged hearts. All these defects contribute to pathogenesis during age-related heart failure [25]. In addition, during cardiac ageing, defective renewal mechanisms for various cellular constituents preclude the clearance of damaged biomolecules and senescent organelles. For example, accumulation of lipofuscin and degenerated mitochondria is evident in aged cardiac muscles. It should be noted that lipofuscin is considered to be one of the age-associated pigments and is found in skin, neurons, and cardiomyocytes [26][27]. These pigment granules are mainly composed of lipid material and highly oxidized proteins that cannot be digested by the ubiquitin-proteasome system and thus accumulate, primarily in lysosomes. The imbalanced lipid metabolism associated with mitochondria, the compromised enzymatic activity present in the lysosomes, and the presence of impaired autophagy pathways, together with inefficient cellular proteostasis, all seem to contribute to the formation of lipofuscin. Accordingly, the accumulation of nondegraded molecules in the lysosomes, including the deposition of lipofuscin, can be used to indicate the presence of age-related damage to both the mitochondria and lysosomes. These findings suggest that defective mitochondria–lysosomal crosstalk is present during cardiac ageing.
The most clinical relevant disorder related to lipofuscin accumulation is neuronal ceroid lipofuscinosis, which involves the accumulation of autofluorescent materials in cardiomyocytes [28][29]. Patients with neuronal ceroid lipofuscinosis display various clinical manifestations, including ventricular hypertrophy, sinus node dysfunction, and arrhythmia [29]; interestingly, these phenotypes are very similar to those associated with the age-related cardiac phenotype. Furthermore, a previous human study using cardiac samples from autopsy patients has revealed that the accumulation of myocardial lipofuscin directly reflects the chronological age of the heart rather than human cardiac pathology [27].
Intriguingly, our studies have shown that Cisd2 can delay cardiac ageing and this is associated with reduced lipofuscin accumulation and preserved myocardial ultrastructure. During the cardiac aging of WT mice, there is a significant increase in lipofuscin accumulation in myocardial tissues. Intriguingly, a high level of Cisd2 reduces lipofuscin accumulation in Cisd2TG mice, which form our long-lived mouse model. Conversely, Cisd2 deficiency significantly elevates lipofuscin accumulation in Cisd2KO mice, which form an accelerated aging mouse model (Figure 1A,B). Moreover, transmission electron microscopy (TEM) analysis has revealed that a high level of Cisd2 expression is able to diminish the accumulation of lipofuscin in cardiomyocytes, to preserve the structural integrity of myofibrils while at the same time ameliorating age-related damage to the ER and mitochondria (Figure 1C,D).
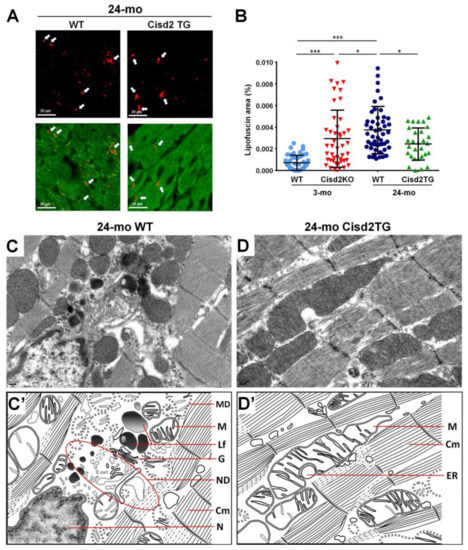
Figure 1. Cisd2 delays cardiac ageing as revealed by reduced lipofuscin accumulation and preserved myocardial ultrastructure. (A) Lipofuscin deposits can be clearly identified in cardiac tissues by autofluorescence detected with 330–380 nm excitation light and 420 nm barrier filter of a confocal fluorescence microscope. The red color indicates lipofuscin and green color indicates myocardia. (B) There is a significant increase of lipofuscin accumulation during cardiac ageing of WT mice. Intriguingly, a high level of Cisd2 reduces lipofuscin accumulation in Cisd2TG mice, while Cisd2 deficiency significantly elevates lipofuscin accumulation in Cisd2KO mice. The areas of lipofuscin accumulation and the myocardia are measured using Fiji/ImageJ 1.52e (National Institutes of Health, Bethesda, MD, USA). Ten fields per subject are observed at 20× objective magnification, and the lipofuscin accumulation (lipofuscin deposit area/myocardial area) was calculated. * p < 0.05, ** p < 0.01, *** p < 0.001. (C,D) Transmission electron microscopy (TEM) analysis for the left ventricle of heart. In 24-mo WT mice (C), age-associated mitochondria degeneration, lipofuscin accumulation, and necrotic debris of degenerative myofibril and organelles are detectable and easily identified. (D) In 24-mo Cisd2TG mice, relatively normal ultrastructures, namely intact mitochondria, ER, and cardiac myofibrils, are found. (C′) (D′) Schematic presentation for ultrastructure of left ventricle shown in (C) and (D), respectively. MD: mitochondrial degeneration, Lf: lipofuscin, ND: necrotic debris of degenerative myofibril and organelles, G: Golgi apparatus, M: mitochondria, Cm: cardiac myofibril, ER: endoplasmic reticulum, N: nucleus.
3. Cisd2 Deficiency Causes Dysregulation of Intracellular Ca2+ Homeostasis
3.1. Mitochondria and Cellular Ca2+ Homeostasis
Calcium signaling plays a crucial role in many molecular processes and a range of cellular functions. Accumulating evidence indicates that Ca2+ strictly controls cellular senescence [6]. An elevation of intracellular Ca2+ levels has been observed in response to different types of stresses. Mitochondria, equipped with Ca2+ uniporters and a negative membrane potential (about −180 mV), are able to function as a potent Ca2+ buffering organelle when there is an elevation in intracellular Ca2+ concentration ([Ca2+]i) [30]. Furthermore, mitochondrial Ca2+ signaling increases when an advanced age is reached [31]. The ER and mitochondria are the two major organelles that govern the intracellular storage, sequestration, and release of [Ca2+]i [32][33]. An elevation in [Ca2+]i is usually attributable to a Ca2+ influx from the extracellular milieu via plasma membrane Ca2+ channels or from intracellular stores of Ca2+ present in the ER [6]. Any release of Ca2+ from ER elevates the cytosolic Ca2+ and this occurs in combination with ever larger cycles of mitochondrial Ca2+ uptake [34]. These increases in mitochondrial Ca2+ concentration leads to a drop in mitochondrial membrane potential, which in turn enhances the production of ROS. As a result, oxidative stress increases further, triggering senescence [6], as well as organ dysfunction, during the aging process [31]. Mitochondrial Ca2+ uniporter (MCU) and H+/Ca2+ exchanger LETM1 are the two main transporters responsible for mitochondrial Ca2+ uptake [35][36]; on the other hand, NCX (Na+/Ca2+ exchanger) and H+/Ca2+ exchanger are the two transporters that export Ca2+ back to cytoplasm [30][37].
3.2. ER-Mitochondrial Ca2+ Dysregulation and Cardiac Ageing
The ER, which consists of a spatially extended membranous network, is often positioned in close proximity to other cellular organelles and these areas form membrane contact sites [38]. The sites of physical interaction and communication between the ER and mitochondria, namely the mitochondria-associated ER membranes (MAMs), represents a platform that is fundamentally important to the modulation of Ca2+ homeostasis, autophagy, apoptosis, lipid metabolism, metabolic diseases, and tumor growth; these events are mediated via the exchange of lipids and Ca2+ ions [5][16][39][40][41][42]. The conserved structures of MAMs found across eukaryotic phyla are key determinants of cell survival and their functionality, which involves the bidirectional trafficking of factors between the two organelles [43][44]. This means that MAMs are dynamic structures that are sensitive to the physiological conditions within cells [16].
Four major functions have been identified for MAMs. These are (a) control of Ca2+ signaling, (b) regulation of mitochondrial division, (c) accommodation of lipid biosynthesis, and (d) coordination of the dynamic interactions between mitochondria and the ER [44]. Importantly, MAMs provide an efficient way for Ca2+ traffic to take place between the ER and mitochondria and this allows the creation of a higher Ca2+ concentration close to the MAMs compared to the surrounding cytoplasm. The Ca2+ released from the ER via the MAMs to the mitochondria means that this high local concentration of Ca2+ is able to activate various Ca2+-dependent processes. These include the tricarboxylic acid (TCA) cycle, oxidative phosphorylation, the regulation of ROS signaling, cell death, and the stimulation of mitochondrial division [14][44][45][46]. A functional protein complex is essential for the tethering of ER-mitochondria and for maintaining the MAM structure; this complex is formed via inositol 1,4,5-trisphosphate receptors (IP3Rs) at ER, voltage dependent anion-selective channel (VDAC) protein in the mitochondrion, and the chaperone 75 kDa glucose regulated protein (GRP75) [14]. In cardiac and skeletal muscle, the Ca2+ channel in the ER, namely the ryanodine receptor (RYR), also allows Ca2+ traffic from ER to mitochondria close to the MAMs [47]. Many other tether complexes of MAMs had also been reported [38]. Interestingly, when the hearts of Mfn2KO mice are examined, the contact lengths of MAMs are reduced by 30%; this is accompanied by an increased level of ROS, and reduced cardiac contractility [48]. Using the norepinephrine-induced cardiac hypertrophic model, it has been shown that the distance between the ER and mitochondria is increased and that this is accompanied by impaired Ca2+ trafficking between the two organelles [49]. Indeed, alterations in the contact length and gap distance of MAMs had been reported to dysregulate Ca2+ signaling and affect cardiac metabolism, thereby accelerating cardiac ageing and promoting the development of cardiac hypertrophy, as well as heart failure [50].
3.3. Cisd2 Maintains Ca2+ Homeostasis
Cisd2 is mainly located in the mitochondrial outer membrane and the ER. In particular, Cisd2 is highly enriched in the MAMs. Previous studies by our group and other researchers have revealed that the intracellular localization and binding partners of Cisd2 vary with the cell type and/or with the experimental conditions [51]. This scenario helps to explain why different proteins have been found to interact with Cisd2 under a variety of circumstances [18][19][21][22][23][52][53]. For example, in the skeletal muscle, Cisd2 appears to associate with Bcl-2/Bcl-XL and IP3R when participating in the regulation of the ER Ca2+ store. Loss of Cisd2 in myocytes results in a dysregulation of Ca2+ homeostasis that is accompanied by augmented autophagy and the presence of degenerated ER [52][53]. On the other hand, in the liver, Cisd2 interacts directly with Serca2b, which is a Ca2+-pump that transports Ca2+ from the cytosol into the ER. Furthermore, investigations have revealed that Cisd2 directly binds to Serca2a in cardiomyocytes. In both cases, this allows there to be control of Ca2+ pump activity by these two proteins via modulation of their oxidative modification. This in turn regulates ER Ca2+ uptake by Serca2a and Serca2b, which then sustains Ca2+ homeostasis in the heart and liver, respectively [18][19]. Finally, in adipocytes, our research has revealed that Cisd2 interacts directly with Gimap5 (GTPase of immune-associated protein 5) on the mitochondrial and ER membranes in order to modulate the Ca2+ buffering capacity of mitochondria, thereby maintaining the intracellular Ca2+ homeostasis of adipocytes. Loss of Cisd2 increases the cytosolic level of Ca2+, and induces Ca2+-calcineurin-dependent signaling that inhibits adipogenesis [21].
Mechanistically, one important question is how Cisd2 modulates intracellular Ca2+ homeostasis via its interaction with its partner proteins. Here we use Serca2 as an example to illustrate the potential molecular mechanism behind the regulation of Ca2+ via Serca2a in the cardiomyocytes and Serca2b in the hepatocytes [18][19]. Three possibilities may explain how the redox status of Serca2a and Serca2b is affected by Cisd2. First, the only defined functional domain of the Cisd2 protein is the protein’s CDGSH domain, which binds a redox-active 2Fe-2S cluster and is oriented toward the cytosol. It seems likely that Cisd2 directly interacts with Serca2 to help maintain Serca2b in a reduced state via the redox capacity of the CDGSH domain present on Cisd2. Second, Cisd2 may cover a part of Serca2 via protein–protein interaction, thus reducing the accessibility of certain tyrosines and cysteines present on Serca2 to attack by enzymes; as a consequence, this may prevent Serca2 from undergoing irreversible oxidative modification (cysteine sulfonation and tyrosine nitration) during oxidative stress. Finally, the redox-active CDGSH domain of Cisd2 may contribute to the maintenance of the general redox status of the cell. This will act to reduce the overall oxidative stress in the cell and indirectly protect Serca2 activity from ROS-mediated and/or RNS-mediated protein oxidation that can bring about a reduction in the protein’s Ca2+ pumping activity. These possibilities are not exclusive, but rather all may contribute to maintaining the redox homeostasis of the Serca2b protein [18].
3.4. Cisd2 Protects the Interfibrillar and Subsarcolemmal Mitochondria from Age-Related Damage
Mitochondria can be divided into three distinct populations based on their location and proximity to Ca2+ release sites [54]. These are, firstly, perinuclear mitochondria (PNMs), which are arranged in clusters and located adjacent to the nucleus; their function is presumably associated with transcription. Secondly, there are subsarcolemmal mitochondria (SSMs), which are also arranged in clusters, but are located just beneath the sarcolemma; their function is presumably associated with the functioning of ion channels and various signaling pathways. Thirdly, there are the interfibrillar mitochondria (IFMs), which are arranged in rows alongside the myofibrils and are located very close to the Ca2+ release sites of the SR; their function is presumably to generate ATP that then contributes to the contractile function of myofibrils, as well as to participate in the Ca2+ signaling within cardiomyocytes [55].
The IFMs are probably the most susceptible mitochondria to age-related damage. Previous studies have revealed that, during the ageing of cardiomyocytes, there is an increase in mitochondrial ROS and a decrease in antioxidant capacity that leads to an increased accumulation of oxidative damage with the DNA [56]; notably this is almost exclusively associated with the IFMs [57]. The abundance of IFMs and their physically close association with myofilaments suggests that IFMs provide the energy needed for cardiomyocyte contraction and thus have a higher respiratory capacity than SSMs [58]. Therefore, any loss of IFMs is likely to adversely affect muscle contractility by limiting the ATP production required by myosin ATPases. Moreover, the increased ROS level associated with aged IFMs is likely to exacerbate mitochondria-derived oxidant injury to the myofilaments; this in turn will lead to increased fibrosis and/or fiber disarrangement, thereby decreasing the contractile capacity of the heart [59]. Additionally, a previous study has shown that decreased mitophagy is accompanied by mitochondrial fragmentation in aging hearts [60]. Intriguingly, these ultrastructural alterations to the IFMs, namely the presence of fragmented mitochondria, the appearance of disarranged and degenerative myofibrils, and the presence of myofibril/organelle necrotic debris, seem to be absent in 24-mo old Cisd2TG mice, which is not the case in naturally aged 24-mo old WT mice (Figure 1C,D).
The function of SSMs is presumably related to ATP production that allows electrolyte and protein transport across the sarcolemma [58]. The exclusive localization of Connexin 43, which is the major component of gap junctions for cell–cell communication, within the SSMs further pinpoints their important role in electrical conductance within the heart [61]. In the naturally aged (24-mo old WT) and prematurely aged (3-mo old Cisd2KO) mice, the SSMs have largely disappeared with only a few degenerating SSMs remaining detectable. This seems to explain the presence of the age-related cardiac conductance abnormalities that occur during ageing (Figure 2A). Remarkably, in the long-lived mice (24-mo old Cisd2TG), a persistently higher level of Cisd2 is able to eliminate age-related damage to the SSMs in the cardiomyocytes; the amount and structural integrity of SSM in the 24-mo old Cisd2TG is similar to that of 3-mo old young WT mice (Figure 2A). This result is consistent with our previous study [19] wherein a higher level of Cisd2 was found to preserve functional mitochondria. This will, in turn, provide a stable MAM microenvironment that ensures efficient communication between the ER and mitochondria, thereby maintaining intracellular Ca2+ homeostasis in the cardiomyocytes. This means that any age-related functional decline of an aging heart, namely changes to mechanical contractility and electrical conductance, are attenuated by the presence of the Cisd2 protein.

Figure 2. Cisd2 preserves the integrity of subsarcolemmal mitochondria and maintains the ultrastructure of mitochondria-associated ER membrane (MAM). (A) Cisd2 preserves the integrity of subsarcolemmal mitochondria and cardiac myofibrils of left ventricle. In 3-mo WT mice, there is an abundance of normal subsarcolemmal mitochondria and normal cardiac myofibrils (A-1 and A-1′). In 3-mo Cisd2KO mice (prematurely aged), notably, a low density of subsarcolemmal mitochondria, and degeneration of mitochondria and myofibrils are found (A-2 and A-2′). In 24-mo WT mice (naturally aged), similar ultrastructural damages are found as those observed in the 3-mo Cisd2KO mice (A-3 and A-3′). Remarkably, in 24-mo Cisd2TG mice, a high level of Cisd2 preserves the density and integrity of subsarcolemmal mitochondria as well as maintains the ultrastructure of cardiac myofibrils (A-4 and A-4′). (A-1′) to (A-4′) are schematic presentations for ultrastructure of left ventricle shown in (A-1) to (A-4). SL: sarcolemma, IS: intercellular space, M: mitochondria, Cm: cardiac myofibril, ICD: intercalated disc, MD: mitochondrial degeneration, Cmd: cardiac myofibril degeneration. (B) Cisd2 is essential to maintaining the integrity of MAM. In 3-mo WT mice, at MAM, mitochondria closely attach to ER (B-1 and B-1′). In 3-mo Cisd2KO mice, notably, a longer distance between mitochondria and ER is found; in addition, mitochondria degeneration is also found (B-2 and B-2′). In 24-mo WT mice, a longer distance between mitochondria and ER is found; this is similar to that observed in the 3-mo Cisd2KO mice (B-3 and B-3′). Intriguingly, in 24-mo Cisd2TG mice, a high level of Cisd2 maintains a relatively normal MAM, where the mitochondria closely attach to the ER (B-4 and B-4′). (B-1′) to (B-4′) are schematic presentations for the ultrastructure of left ventricle shown in (B-1) to (B-4). The inset shows a higher magnification to provide a better illustration for MAM. The inset shows a higher magnification of the selected area (yellow squares) of the middle panel to provide a better illustration for MAM.
3.5. Cisd2 Maintains the MAMs during Cardiac Ageing
Normal functioning of MAMs is associated with there being an appropriate width of the cleft between the mitochondrial outer membrane (OMM) and the ER [62]. Certain structural arrangements within the space that separates the ER and mitochondria are able to support an optimal Ca2+ transfer via the Ca2+ transport channels [46]. Thus, there is an optimal distance of ≈30 nm between ER and mitochondria that allows effective Ca2+ transfer from ER, through the Ca2+ channel IP3R located in ER, to the mitochondria, through the Ca2+ channel MCU located in mitochondria. This Ca2+ transfer contributes to the generation of physiologically relevant cytosolic Ca2+ oscillations [63]. In addition, a previous study has defined the role of this microenvironment as being the key compartment driving cytosolic Ca2+ oscillations. The microenvironment between the ER and mitochondria plays a critical role in Ca2+ dynamics by modulating the activity of IP3Rs and allowing the shuttling of Ca2+ between the ER and mitochondria [64]. A disruption of ER-mitochondria integrity and communication and a consequential dysregulation of Ca2+ homeostasis have been linked with pathogenesis, aberrant metabolism [38][42], neurodegenerative disease [38], a decreased lifespan [65], and ageing [15][41].
If the cleft between OMM and ER is too narrow, this causes various abnormalities. For example, under ER stress, MAMs become closer to the mitochondria, with a 25% decrease in distance and a 60% increase in contact length between the two organelles [39]. This results in an enhanced transport of Ca2+ from the ER to the adjacent mitochondrial network, which stimulates oxidative metabolism and induces the apoptotic program [16][34]. Additionally, during long-term cultured neuron, which is a neuronal senescence model, an increased Ca2+ transport from the ER to mitochondria has been associated with an upregulation of the MCU [66]. Thus, it seems that increased Ca2+ transfer to mitochondria and an accumulation of Ca2+ within the mitochondria seems to serve as one of the mechanisms underlying the loss of mitochondrial membrane potential and the induction of cell senescence [67].
If the cleft between the OMM and ER is too wide, this also causes problems. A previous study has revealed that abnormally high cytosolic Ca2+ develops when the distance is greater than the optimum [63]. Remarkably, our studies have revealed that Cisd2 deficiency or a decreased expression of Cisd2 to less than 50%, namely haploinsufficient, disrupts Ca2+ homeostasis and causes elevated cytosolic Ca2+ levels. This Cisd2-mediated Ca2+ dysregulation is accompanied by abnormal dilation of the ER and a breakdown of the mitochondrial outer membrane [17][18][19] in many different tissues and organs, including brown adipose tissue [68], skeletal muscles [22][53], during neurodegenerative Alzheimer’s disease [23], and during cardiac dysfunction [19].
In naturally aged WT mice at 24-mo old, in which the level of Cisd2 has decreased to less than 50% in the heart, and in prematurely aged Cisd2KO mice at 3-mo old, in which Cisd2 expression is completely eliminated in the heart, the MAM gap distance is obviously increased (Figure 2B); this seems to provide an explanation for the differences in cardiac metabolism and Ca2+ signaling that occur with these two mouse models [19]. Intriguingly, in the long-lived Cisd2TG mice at 24-mo old, in which Cisd2 is 2-fold higher in the heart, the compactness and proximity of the ER and mitochondria appears to be well preserved (Figure 2B). This probably ensures that there is correct contact of the mitochondria with the MAM, which then allows normal Ca2+ trafficking between these two organelles to be maintained. The cardiac phenotypes of Cisd2KO mice and Cisd2TG mice are summarized in Table 1.
Table 1. Compare the cardiac phenotypes of WT, Cisd2KO and Cisd2TG mice.
| Age | Young Age (3 Months Old) |
Old Age (24–26 Months Old) |
||
|---|---|---|---|---|
| Genotype | WT | Cisd2KO | WT | Cisd2TG |
| Genetic modification | - | Conventional Cisd2 KO | - | Carrying 4 copies of Cisd2 gene |
| Cisd2 protein level | 100% | 0% | 50% | 100-130% |
| Cardiac mechanical function (Ejection Fraction) | ~85% | ~80% (~70% at 6 months old) |
~60% | ~75% |
| Cardiac electrical function | Sinus rhythm |
|
|
|
| Histopathology | Rare lipofuscin |
|
|
|
| Ultrastructure of cardiomyocyte | Normal |
|
|
|
| Calcium homeostasis | Normal |
|
|
|
| Mito 4 function (OCR) | 100% | 61% | Not determined | Not determined |
| Oxidative stress (fold increase) | 1.0 | 2.3 | 2.6 | 1.2 |
1 APC, Atrial Premature Contractions; 2 Cyt, Cytosole; 3 MAM, Mitochondria-Associated ER Membrane; 4 Mito, Mitochondria; 5 SR, Sarcoplasmic Reticulum; 6 VPCs, Ventricular Premature Contractions.
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hastings, M.H.; Rhee, J.; Trager, L.E.; Roh, J.D.; Rosenzweig, A. Targeting age-related pathways in heart failure. Circ. Res. 2020, 126, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, D.; Brundel, B.; Wiersma, M. Imbalance of ER and mitochondria interactions: Prelude to cardiac ageing and disease? Cells 2019, 8, 1617. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef]
- Martin, N.; Bernard, D. Calcium signaling and cellular senescence. Cell Calcium 2018, 70, 16–23. [Google Scholar] [CrossRef]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid. Med. Cell Longev. 2019, 2019, 9825061. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Nathania, M.; Hollingsworth, K.G.; Bates, M.; Eggett, C.; Trenell, M.I.; Velicki, L.; Seferovic, P.M.; MacGowan, G.A.; Turnbull, D.M.; Jakovljevic, D.G. Impact of age on the association between cardiac high-energy phosphate metabolism and cardiac power in women. Heart 2018, 104, 111–118. [Google Scholar] [CrossRef]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) redox control and mitochondrial metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Silva-Palacios, A.; Zazueta, C.; Pedraza-Chaverri, J. ER membranes associated with mitochondria: Possible therapeutic targets in heart-associated diseases. Pharmacol. Res. 2020, 156, 104758. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Janikiewicz, J.; Szymanski, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszynski, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Chen, Y.F.; Kao, C.H.; Chen, Y.T.; Wang, C.H.; Wu, C.Y.; Tsai, C.Y.; Liu, F.C.; Yang, C.W.; Wei, Y.H.; Hsu, M.T. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef]
- Shen, Z.Q.; Chen, Y.F.; Chen, J.R.; Jou, Y.S.; Wu, P.C.; Kao, C.H.; Wang, C.H.; Huang, Y.L.; Chen, C.F.; Huang, T.S. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef]
- Yeh, C.H.; Shen, Z.Q.; Hsiung, S.Y.; Wu, P.C.; Teng, Y.C.; Chou, Y.J.; Fang, S.W.; Chen, C.F.; Yan, Y.T.; Kao, L.S. Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 2019, 17, e3000508. [Google Scholar] [CrossRef]
- Chen, Y.F.; Kao, C.H.; Kirby, R.; Tsai, T.F. Cisd2 mediates mitochondrial integrity and life span in mammals. Autophagy 2009, 5, 1043–1045. [Google Scholar] [CrossRef]
- Wang, C.H.; Chen, Y.F.; Wu, C.Y.; Wu, P.C.; Huang, Y.L.; Kao, C.H.; Lin, C.H.; Kao, L.S.; Tsai, T.F.; Wei, Y.H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Shen, Z.Q.; Wu, C.Y.; Teng, Y.C.; Liao, C.C.; Kao, C.H.; Chen, L.K.; Lin, C.H.; Tsai, T.F. Comparative proteomic profiling reveals a role for Cisd2 in skeletal muscle aging. Aging Cell 2018, 17, e12705. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Chou, T.Y.; Lin, I.H.; Chen, C.G.; Kao, C.H.; Huang, G.J.; Chen, L.K.; Wang, P.N.; Lin, C.P.; Tsai, T.F. Upregulation of Cisd2 attenuates Alzheimer’s-related neuronal loss in mice. J. Pathol. 2020, 250, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: Mechanisms and therapeutic opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Garcia, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef]
- Kakimoto, Y.; Okada, C.; Kawabe, N.; Sasaki, A.; Tsukamoto, H.; Nagao, R.; Osawa, M. Myocardial lipofuscin accumulation in ageing and sudden cardiac death. Sci. Rep. 2019, 9, 3304. [Google Scholar] [CrossRef]
- Rietdorf, K.; Coode, E.E.; Schulz, A.; Wibbeler, E.; Bootman, M.D.; Ostergaard, J.R. Cardiac pathology in neuronal ceroid lipofuscinoses (NCL): More than a mere co-morbidity. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165643. [Google Scholar] [CrossRef]
- Østergaard, J.R.; Rasmussen, T.B.; Mølgaard, H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011, 76, 1245–1251. [Google Scholar] [CrossRef]
- Olson, M.L.; Chalmers, S.; McCarron, J.G. Mitochondrial organization and Ca2+ uptake. Biochem. Soc. Trans. 2012, 40, 158–167. [Google Scholar] [CrossRef]
- Lopes, G.S.; Ferreira, A.T.; Oshiro, M.E.; Vladimirova, I.; Jurkiewicz, N.H.; Jurkiewicz, A.; Smaili, S.S. Aging-related changes of intracellular Ca2+ stores and contractile response of intestinal smooth muscle. Exp. Gerontol. 2006, 41, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Segal, S.S. Impact of aging on calcium signaling and membrane potential in endothelium of resistance arteries: A role for mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Nobili, A.; Krashia, P.; D’Amelio, M. Cisd2: A promising new target in Alzheimer’s disease. J. Pathol. 2020, 251, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef]
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 2013, 1832, 495–508. [Google Scholar] [CrossRef]
- Missiroli, S.; Danese, A.; Iannitti, T.; Patergnani, S.; Perrone, M.; Previati, M.; Giorgi, C.; Pinton, P. Endoplasmic reticulum-mitochondria Ca(2+) crosstalk in the control of the tumor cell fate. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 858–864. [Google Scholar] [CrossRef]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Periwal, V. A mathematical model of calcium dynamics: Obesity and mitochondria-associated ER membranes. PLoS Comput. Biol. 2019, 15, e1006661. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum–mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gibhardt, C.S.; Will, T.; Stanisz, H.; Korbel, C.; Mitkovski, M.; Stejerean, I.; Cappello, S.; Pacheu-Grau, D.; Dudek, J.; et al. Redox signals at the ER-mitochondria interface control melanoma progression. EMBO J. 2019, 38, e100871. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Csordas, G.; Weaver, D.; Hajnoczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Dorn, G.W., II; Song, M.; Walsh, K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J. Mol. Cell Cardiol. 2015, 78, 123–128. [Google Scholar] [CrossRef]
- Gutiérrez, T.; Parra, V.; Troncoso, R.; Pennanen, C.; Contreras-Ferrat, A.; Vasquez-Trincado, C.; Morales, P.E.; Lopez-Crisosto, C.; Sotomayor-Flores, C.; Chiong, M.; et al. Alteration in mitochondrial Ca2+ uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun. Signal. 2014, 12, 68. [Google Scholar] [CrossRef]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 2017, 14, 342–360. [Google Scholar] [CrossRef]
- Wang, C.H.; Kao, C.H.; Chen, Y.F.; Wei, Y.H.; Tsai, T.F. Cisd2 mediates lifespan: Is there an interconnection among Ca(2)(+) homeostasis, autophagy, and lifespan? Free Radic. Res. 2014, 48, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Nguyen, M.; Bourdon, J.; Risse, P.-A.; Martin, J.; Danialou, G.; Rizzuto, R.; Petrof, B.J.; Shore, G.C. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012, 21, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.P.; Zheng, M. Mitochondrial dynamics and inter-mitochondrial communication in the heart. Arch. Biochem. Biophys. 2019, 663, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shigeno, E.T.; Morrow, J.D.; Cox, B.; Rocha, A.E.; Frei, B.; Hagen, T.M. Oxidative stress in the aging rat heart is reversed by dietary supplementation with (R)-(alpha)-lipoic acid. FASEB J. 2001, 15, 700–706. [Google Scholar] [CrossRef]
- Suh, J.H.; Heath, S.H.; Hagen, T.M. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic. Biol. Med. 2003, 35, 1064–1072. [Google Scholar] [CrossRef]
- Ulasova, E.; Gladden, J.D.; Chen, Y.; Zheng, J.; Pat, B.; Bradley, W.; Powell, P.; Zmijewski, J.W.; Zelickson, B.R.; Ballinger, S.W.; et al. Loss of interstitial collagen causes structural and functional alterations of cardiomyocyte subsarcolemmal mitochondria in acute volume overload. J. Mol. Cell Cardiol. 2011, 50, 147–156. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef]
- Stotland, A.; Gottlieb, R.A. α-MHC MitoTimer mouse: In vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J. Mol. Cell Cardiol. 2016, 90, 53–58. [Google Scholar] [CrossRef]
- Boengler, K.; Stahlhofen, S.; van de Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Li, L.; Shuai, J. Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci. Rep. 2015, 5, 7984. [Google Scholar] [CrossRef]
- Moshkforoush, A.; Ashenagar, B.; Tsoukias, N.M.; Alevriadou, B.R. Modeling the role of endoplasmic reticulum-mitochondria microdomains in calcium dynamics. Sci. Rep. 2019, 9, 17072. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca(2+) cross talk and loss of store-operated Ca(2+) entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 2016, 1863, 2637–2649. [Google Scholar] [CrossRef]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calve, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef]
- Gao, P.; Jiang, Y.; Wu, H.; Sun, F.; Li, Y.; He, H.; Wang, B.; Lu, Z.; Hu, Y.; Wei, X.; et al. Inhibition of Mitochondrial Calcium Overload by SIRT3 Prevents Obesity- or Age-Related Whitening of Brown Adipose Tissue. Diabetes 2020, 69, 165–180. [Google Scholar] [CrossRef]


