 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kenjiro Ono | + 2264 word(s) | 2264 | 2020-02-03 09:28:38 | | | |
| 2 | Nicole Yin | -115 word(s) | 2149 | 2020-10-23 09:45:06 | | |
Video Upload Options
Worldwide, Alzheimer's disease (AD) is the most common age-related neurodegenerative disease and is characterized by unique pathological hallmarks in the brain, including plaques composed of amyloid β-protein (Aβ) and neurofibrillary tangles of tau protein. Genetic studies, biochemical data, and animal models have suggested that Aβ is responsible for the pathogenesis of AD (i.e., the amyloid hypothesis). Indeed, Aβ molecules tend to aggregate, forming oligomers, protofibrils, and mature fibrils. However, while these Aβ species form amyloid plaques of the type implicated in AD neurodegeneration, recent clinical trials designed to reduce the production of Aβ and/or the plaque burden have not demonstrated clinical efficacy. In addition, recent studies using synthetic Aβ peptides, cell culture models, Arctic transgenic mice, and human samples of AD brain tissues have suggested that the pre-fibrillar forms of Aβ, particularly Aβ protofibrils, may be the most critical species, compared with extracellular fibrillar forms. We recently reported that protofibrils of Aβ1-42 disturbed membrane integrity by inducing reactive oxygen species generation and lipid peroxidation, resulting in decreased membrane fluidity, intracellular calcium dysregulation, depolarization, and synaptic toxicity. Therefore, the therapeutic reduction of protofibrils may prevent the progression of AD by ameliorating neuronal damage and cognitive dysfunction through multiple mechanisms.
1. The Discovery of PFs and Their Role in AD Pathogenesis
PFs were first described by Teplow and colleagues in 1997[1]. Using a size exclusion chromatography (SEC) system and the synthetic Aβ1-42 peptide, they found a peak representing a large (> 100 kDa) soluble species before the peak of the LMW-Aβ (mainly monomer)[1]. Using electron microscopy (EM), they further revealed that this peak contained predominantly curved fibrils, with a diameter of ~5 nm and a length of up to 200 nm, which they termed PFs[1]. Subsequently, the authors elucidated that the PFs were composed primarily of β-sheets and partially random coils and α-helices in a secondary structure[1]. In the same year, using atomic force microscopy (AFM), Lansbury’s group found the existence of a metastable intermediate species, which was termed Aβ PF[2]. Many data have shown that LMW-Aβ oligomers are on-pathway to fibril formation, while HMW-Aβ oligomers such as PFs are off-pathway[2][3][4][5]. Although the PF-to-fibril transition, characterized by PF elongation, was very slow, preformed fibrillar seeds greatly accelerated this conversion[2]. Recently, using a combination of high-speed AFM with thioflavin T assay, EM, and re-injection assays by SEC, we demonstrated that fibril formation from PFs is more difficult than that from LMW-Aβ, suggesting that mature fibrils of Aβ1-42 are primarily formed from LMW-Aβ1-42 and not from PFs[4]. Furthermore, we determined that PFs instead supplied precursors to LMW-Aβ1-42 by their dissociation, suggesting that PFs may not always represent the “on-pathway” of Aβ1-42 aggregation from the monomer to the mature fibrils[4]. Kodali and Wetzel mentioned that, although Aβ1-40 PFs can grow by monomer addition, their rate of growth is lower than that of mature fibrils. Additionally, while Aβ1-40 monomer was able to support the extension of mature fibrils at low concentrations of, Aβ1-40 PFs exhibited no extension[3]. They suggested another terminology, “curvilinear fibrils”, for the description of off-pathway PFs instead of PFs as on-pathway precursors of fibrils[3]. It was recently revealed that curvilinear fibrils inhibit fibril formation not only by slowing fibril nucleation and elongation, but also by actively disrupting either process based on combined thioflavin kinetics and AFM imaging data[6]. On the other hand, Iwatsubo’s group showed that Aβ1-42 PF injection induced Aβ deposition in the brains of A7 mice overexpressing human APP695 and harboring the K670N, M671L, and T714I familial AD neuronal mutations, suggesting that Aβ PFs may act as a seed for Aβ aggregation in vivo[7]. The injection of Aβ PFs mixed with apoE3 significantly attenuated Aβ deposition, whereas apoE4 did not, suggesting that the suppressive effect of apoE3 on the structural conversion of Aβ PFs to fibrils is stronger than that of apoE4, thereby impeding Aβ deposition in vivo[7].
2. PFs Are Primary Toxins in AD
The solubility and diffusible nature of soluble oligomers may render them more effective in terms of intra- and extra-cellular interactions and engaging microglial receptors compared with mature insoluble fibrils. Indeed, it has been demonstrated that astrocytes engulf large amounts of accumulated, rather than digested, Aβ1-42 PFs. This intracellular storage of Aβ1-42 results in severe astrocytic endosomal/lysosomal defects and the secretion of extracellular vesicles containing N-truncated, neurotoxic Aβ[8]. Aβ1-42 PFs have also been shown to induce an inflammatory process through microglial activation[9] and initiate Toll‐like receptor (TLR) signaling (Figure 1)[10]. In addition, these PFs are preferentially internalized by microglia[11]. Furthermore, it has been reported that Aβ1-42 PFs are more effective at inducing microglial tumor necrosis factor α (TNFα) production in BV-2 and primary murine microglia in vitro than monomers and mature fibrils. Moreover, PFs of Aβ1-40 exhibit significantly less activity than concentration-matched Aβ1-42[9]. Aβ1-42 PFs also have been shown to trigger a time- and myeloid differentiation protein (MyD) 88-dependent process that generates TNFα and interleukin-1β (IL-1β) mRNA, along with pro and mature forms of the intracellular IL-1β protein[10]. The accumulation of both IL-1β forms has indicated that Aβ1-42 PFs are able to prime and activate the Nod-like receptor (NLR) P3 inflammasome. In this process, Aβ has been shown to elicit a quantized burst of secreted IL-1β which occurs prior to the Aβ priming of the microglia. The IL-1β secretion burst appears to be rapid and not sustained, yet it may be re-initiated with additional Aβ stimulation. These findings indicate multiple modes of IL-1β regulation by Aβ1-42 PFs, including TLR/MyD88-mediated priming, NLRP3 inflammasome activation, and modulation of the IL-1β secretory process, suggesting wide-ranging effects of Aβ on the innate immune response[10].
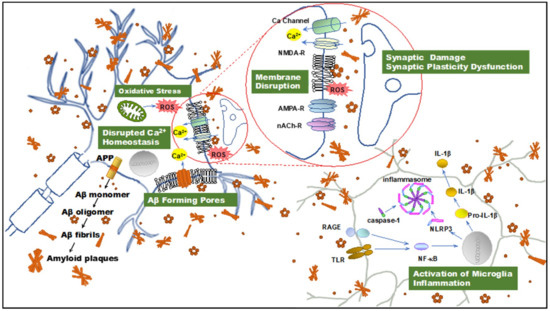
Figure 1. Illustration summarizing amyloid β-protein (Aβ) neurotoxicity. Aβ aggregates induce disruption of cellular homeostasis, which may be the result of inducing or exacerbating membrane disruption, oxidative stress, calcium dysregulation, synaptic plasticity dysfunction, and inflammation. APP: amyloid precursor protein; Aβ: amyloid β-protein; ROS: reactive oxygen species; NMDAR, N-methyl-d-aspartate receptor. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; nAChR, nicotinic acetylcholine receptor; TLR: toll-like receptor; RAGE: receptor for advanced glycation endproducts; NF-κB, nuclear factor κB; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; IL-1β: interleukin-1β.
Recent evidence has suggested that the neuronal cell membrane is the chief site of oligomer-mediated neuronal damage. We recently studied the cellular response to short exposures to PFs using multiple indices of membrane integrity, cytolysis, oxidative stress, and synaptic function. We found that cellular membrane and metabolic integrity were more severely disrupted by PFs of Aβ1-42 than LMW-Aβ1-42, as evidenced by various experimental systems, including cell viability and leakage assays, fluorometric measures of ROS generation, lipid peroxidation assays, and electrophysiological recordings[12]. While our results for lactate dehydrogenase (LDH) and calcein and ethidium homodimer-1 assays reflected cellular membrane damage by PFs of Aβ1-42 to a greater extent than LMW-Aβ1-42, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide metabolism (MTT) and water soluble tetrazolium (WST) assays reflecting mitochondrial enzyme activity, they demonstrated only small differences between the Aβs in different cellular models, including SH-SY5Y cells and a healthy, human-induced pluripotent stem line[12]. From these results, in terms of short-term Aβ1-42 PF treatment, Aβ1-42 PFs may first attack the cell membrane, followed by subsequent damage to the mitochondria, although Aβ1-42 dimers might not be removed clearly in LMW-Aβ1-42 preparation using the above-mentioned SEC method[1] [6]. Next, we found that exposure to PFs of Aβ1-42 in SH-SY5Y cells induces more severe oxidative stress, including greater levels of ROS production and membrane lipid peroxidation, than LMW-Aβ1-42. Indeed, many studies have reported that oxidative stress, which occurs in the presence of a physiological imbalance between ROS generation and antioxidant capacity, is a critical pathogenic mechanism in AD progression[13]. Along with the direct destruction/modification of lipids, DNA, and proteins, the byproducts of lipid peroxidation produced during oxidative stress cause damage to the mitochondria and upregulate tau phosphorylation, which appears essential for NFT formation[14]. In addition, the generation of superoxide by Aβ aggregates may lead to mitochondrial impairment and further induce ROS generation, thereby establishing a positive feedback pathway that ultimately results in cell death[15]. Moreover, Aβ aggregates may directly interact with the mitochondrial respiratory chain, causing metabolic dysfunction and increased ROS production[16]. In our study, the PFs of Aβ1-42 also reduced neuronal membrane fluidity to a significantly greater extent than LMW-Aβ1-42. Thus, we consider the possibility that the effects on membrane fluidity, and the resulting neuronal damage, depend on the specific Aβ conformation[12].
We further demonstrated that short exposures to Aβ1-42 PFs induces higher concentrations of [Ca2+]i than LMW-Aβ1-42, whereas a reduced depolarization-induced [Ca2+]i influx through voltage-dependent Ca2+ channels was observed following longer exposures to Aβ1-42 PFs[12]. These results suggested that PFs may not only directly damage voltage-gated calcium channels for a short time, but also alter the cell membrane environment required for proper channel insertion or gating for longer periods, as evidenced by lipid peroxidation and membrane fluidity measurements[12].
Consistent with the changes observed in [Ca2+]i and the loss of membrane integrity, the application of PFs of Aβ1-42, but not those of LMW-Aβ1-42, also has been shown to depolarize SH-SY5Y cells and significantly reduce membrane input resistance[12]. Bode et al. monitored transmembrane currents during Aβ exposure at the extracellular face of excised membranes from HEK293 cells, and found that annular Aβ1-42 oligomers formed ion channels, whereas Aβ1-40 oligomers and mature fibrils and monomers did not[17]. Drolle et al. used multi-component lipid models to mimic healthy and AD states of neuronal membranes and posited that Aβ1-42 increases lipid membrane roughness and membrane conductance, possibly through pore formation[18]. Taken together, the [Ca2+]i increase evoked by PFs may be due to pore formation and oxidative damage, as well as the suppression of calcium egress and sequestration pathways secondary to metabolic disruption.
We also demonstrated that Aβ1-42 PFs significantly inhibit LTP formation in the mouse hippocampal CA1 subfield[12]. Similarly, it has been reported that PFs induce electrophysiological changes, including rapid increases in the excitatory post synaptic and action potentials, membrane depolarizations in rat cortical neurons[19], and the inhibition of LTP in the rat hippocampus[20]. Excessive ROS accumulation and decreased membrane fluidity are associated with effects on LTP and learning[21][22] . Furthermore, membrane pore formation may also impair cellular and synaptic functions (Figure 1)[23][24].
The small (35kDa) and highly dispersible protein, secreted-frizzled-related protein 1 (SFRP1), regulates transmembrane metalloprotease ADAM10 activity and is essential for the development of tissue homeostasis and constitutive levels of α-secretase in the brain[25]. As a novel player in AD pathogenesis, SFRP1 has been shown to be significantly increased in the brain and cerebrospinal fluid of patients with AD. In addition, SFRP1 has been demonstrated in human AD cases and mouse models to prevent Aβ PF formation by binding to Aβ, suggesting it may be a promising AD therapeutic target[26].
3. Arctic Mutation Causes Aβ PF Formation
Arctic mutation is a pathogenic APP mutation located within the Aβ sequence at codon 693, at which point glutamic acid is substituted for glycine (E693G). In 2001, Lannfelt’s group named the mutation the ‘Arctic’ mutation because the family in which it was detected was from northern Sweden[27]. Affected subjects have clinical features of early AD and plasma levels of both Aβ1-40 and Aβ1-42 are lower in mutation carriers compared with healthy family members. In addition, concentrations of Aβ1-42 were found to be reduced in media from cells transfected with APPE693G[27]. Furthermore, the authors reported that the Arctic Aβ mutation (Aβ40 Arc) causes enhanced the formation of Aβ1-40 PFs in vitro[27]. Subsequently, Lannfelt’s group found that the Arctic mutation significantly accelerated Aβ1-42 PF formation, as well as PF fibrillization[28].
It has been reported that Aβ1-40 Arc inhibits LTP ~100-fold more potently than wild-type Aβ40 when wild-type and Aβ1-40 Arc peptides are injected into the CA1 area in rats intracerebroventricularly. In this study, the isolated soluble fraction that included the PFs of Aβ1-40 Arc after high-speed centrifugation was shown to still retain full LTP inhibitory activity[20]. In a later study, Lord et al. demonstrated that the Arc mutation accelerates early intraneuronal Aβ aggregation and PF formation, followed by plaque formation, in APP transgenic mice with both the Arctic (E693G) and Swedish (K670N, M671L) mutations (tg-APPArcSwe)[29][30]. In addition, cognitive deficits have been shown to occur concomitantly with the formation of intracellular Aβ deposits, but before plaque formation, in transgenic mice[29]. In addition, the levels of PFs in the brain, but not those of total Aβ, have been correlated with spatial learning, which adds further evidence to the theory that soluble PFs are the toxic species[31]. The pool of toxic Aβ species reportedly consists of molecules in the size range of 80 to 500 kDa[32].
References
- Dominic M. Walsh; Aleksey Lomakin; George B. Benedek; Margaret M. Condron; David B. Teplow; Amyloid β-Protein Fibrillogenesis. Journal of Biological Chemistry 1997, 272, 22364-22372, 10.1074/jbc.272.35.22364.
- James D. Harper; Stanislaus S. Wong; Charles M. Lieber; Peter T. Lansbury; Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chemistry & Biology 1997, 4, 119-125, 10.1016/s1074-5521(97)90255-6.
- Kodali, R.; Wetzel, R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 2007, 17, 48–57.
- Watanabe-Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1-42 aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. ]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610.
- Filip Hasecke; Tatiana Miti; Carlos Perez; Jeremy Barton; Daniel Schölzel; Lothar Gremer; Clara S. R. Grüning; Garrett Matthews; Georg Meisl; Tuomas P. J. Knowles; et al.Dieter WillboldPhilipp NeudeckerHenrike HeiseGhanim UllahWolfgang HoyerMartin Muschol Origin of metastable oligomers and their effects on amyloid fibril self-assembly. Chemical Science 2018, 9, 5937-5948, 10.1039/c8sc01479e.
- Yukiko Hori; Tadafumi Hashimoto; Hidetoshi Nomoto; Bradley T. Hyman; Takeshi Iwatsubo; Role of apolipoprotein E in β-amyloidogenesis: Isoform-specific effects on protofibril to fibril conversion of Aβ in vitro and brain Aβ deposition in vivo.. Journal of Biological Chemistry 2018, 293, 7267-7267, 10.1074/jbc.AAC118.003576.
- Sofia Söllvander; Elisabeth Nikitidou; Robin Brolin; Linda Söderberg; Dag Sehlin; Lars Lannfelt; Anna Erlandsson; Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Molecular Neurodegeneration 2016, 11, 1-19, 10.1186/s13024-016-0098-z.
- Geeta S. Paranjape; Lisa K. Gouwens; David C. Osborn; Michael R. Nichols; Isolated Amyloid-β(1–42) Protofibrils, But Not Isolated Fibrils, Are Robust Stimulators of Microglia. ACS Chemical Neuroscience 2012, 3, 302-311, 10.1021/cn2001238.
- Shana E. Terrill-Usery; Michael J. Mohan; Michael R. Nichols; Amyloid-β(1-42) protofibrils stimulate a quantum of secreted IL-1β despite significant intracellular IL-1β accumulation in microglia.. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2014, 1842, 2276-2285, 10.1016/j.bbadis.2014.08.001.
- Lisa K. Gouwens; Nyasha J. Makoni; Victoria A. Rogers; Michael R. Nichols; Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers.. Brain Research 2016, 1648, 485-495, 10.1016/j.brainres.2016.08.016.
- Taro Yasumoto; Yusaku Takamura; Mayumi Tsuji; Takahiro Watanabe‐Nakayama; Keiko Imamura; Haruhisa Inoue; Shiro Nakamura; Tomio Inoue; Atsushi Kimura; Satoshi Yano; et al.Hisao NishijoYuji KiuchiDavid B. TeplowKenjiro Ono High molecular weight amyloid β 1‐42 oligomers induce neurotoxicity via plasma membrane damage. The FASEB Journal 2019, 33, 9220-9234, 10.1096/fj.201900604r.
- Michela Rosini; Elena Simoni; Andrea Milelli; Anna Minarini; Carlo Melchiorre; Oxidative Stress in Alzheimer’s Disease: Are We Connecting the Dots?. Journal of Medicinal Chemistry 2013, 57, 2821-2831, 10.1021/jm400970m.
- Mubeen A. Ansari; Stephen W. Scheff; Oxidative Stress in the Progression of Alzheimer Disease in the Frontal Cortex. Journal of Neuropathology & Experimental Neurology 2010, 69, 155-167, 10.1097/nen.0b013e3181cb5af4.
- Miroslav Pohanka; Alzheimer´s Disease and Oxidative Stress: A Review. Current Medicinal Chemistry 2013, 21, 356-364, 10.2174/09298673113206660258.
- Luis Fernando Hernandez-Zimbron; Jose Luna-Muñoz; Raul Mena; Ricardo Vazquez-Ramirez; Carlos Kubli-Garfias; David H. Cribbs; Karen Manoutcharian; Goar Gevorkian; Amyloid-β Peptide Binds to Cytochrome C Oxidase Subunit 1. PLoS ONE 2012, 7, e42344, 10.1371/journal.pone.0042344.
- David C. Bode; Mark D. Baker; John H. Viles; Ion Channel Formation by Amyloid-β42Oligomers but Not Amyloid-β40in Cellular Membranes. Journal of Biological Chemistry 2016, 292, 1404-1413, 10.1074/jbc.m116.762526.
- Elizabeth Drolle; Alexander Negoda; Keely Hammond; Evgeny Pavlov; Zoya Leonenko; Changes in lipid membranes may trigger amyloid toxicity in Alzheimer's disease. PLoS ONE 2017, 12, e0182194.
- Dean M Hartley; Dominic M Walsh; Chianping P. Ye; Thekla Diehl; Sara Vasquez; Peter M Vassilev; David B. Teplow; Dennis J. Selkoe; Protofibrillar Intermediates of Amyloid β-Protein Induce Acute Electrophysiological Changes and Progressive Neurotoxicity in Cortical Neurons. The Journal of Neuroscience 1999, 19, 8876-8884, 10.1523/jneurosci.19-20-08876.1999.
- Igor Klyubin; Dominic M. Walsh; William K. Cullen; Julia V. Fadeeva; Roger Anwyl; Dennis J. Selkoe; Michael J. Rowan; Soluble Arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivo. European Journal of Neuroscience 2004, 19, 2839-2846, 10.1111/j.1460-9568.2004.03389.x.
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 2011, 14, 2013–2054.
- Schaeffer, E.L.; Bassi, F., Jr.; Gattaz, W.F. Inhibition of phospholipase A2 activity reduces membrane fluidity in rat hippocampus. J. Neural. Transm. (Vienna) 2005, 112, 641–647.
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017, 13, 178–182.
- Berridge, M.J. Calcium signalling in health and disease. Biochem. Biophys. Res. Commun. 2017, 485, 5.
- Ellen Jorissen; Johannes Prox; Christian Bernreuther; Silvio Weber; Ralf Schwanbeck; Lutgarde Serneels; A. Snellinx; Katleen Craessaerts; Amantha Thathiah; Ina Tesseur; et al.Udo BartschGisela WeskampCarl P. BlobelMarkus GlatzelBart De StrooperPaul Saftig The Disintegrin/Metalloproteinase ADAM10 Is Essential for the Establishment of the Brain Cortex. Journal of Neuroscience 2010, 30, 4833-4844, 10.1523/JNEUROSCI.5221-09.2010.
- Pilar Esteve; Javier Rueda-Carrasco; María Inés Mateo; María Jesús Martin-Bermejo; Jonathan Draffin; Guadalupe Pereyra; África Sandonís; Inmaculada Crespo; Inmaculada Moreno; Ester Aso; et al.Paula Garcia-EsparciaEstrella Gomez-TortosaAlberto RábanoJuan ForteaDaniel AlcoleaAlberto LleoMichael T. HenekaJosé M. ValpuestaJosé A. EstebanIsidro FerrerMercedes DomínguezPaola Bovolenta Elevated levels of Secreted-Frizzled-Related-Protein 1 contribute to Alzheimer’s disease pathogenesis. Nature Neuroscience 2019, 22, 1258-1268, 10.1038/s41593-019-0432-1.
- Camilla Nilsberth; Anita Westlind-Danielsson; Christopher B. Eckman; Margaret M. Condron; Karin Axelman; Charlotte Forsell; Charlotte Stenh; Johan Luthman; David B. Teplow; Steven G. Younkin; et al.Jan NäslundLars Lannfelt The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nature Neuroscience 2001, 4, 887-893, 10.1038/nn0901-887.
- Ann-Sofi Johansson; Fredrik Berglind-Dehlin; Göran Karlsson; Katarina Edwards; Pär Gellerfors; Lars Lannfelt; Physiochemical characterization of the Alzheimer's disease-related peptides Aβ1-42Arctic and Aβ1-42wt. FEBS Journal 2006, 273, 2618-2630, 10.1111/j.1742-4658.2006.05263.x.
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol. Aging 2007, 28, 1297–1306.
- Lord, A.; Kalimo, H.; Eckman, C.; Zhang, X.Q.; Lannfelt, L.; Nilsson, L.N. The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 2006, 27, 67–77.
- Anna Lord; Hillevi Englund; Linda Söderberg; Stina Tucker; Fredrik Clausen; Lars Hillered; Marcia Gordon; Dave Morgan; Lars Lannfelt; Frida Ekholm Pettersson; et al.Lars N. G. Nilsson Amyloid-β protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS Journal 2009, 276, 995-1006, 10.1111/j.1742-4658.2008.06836.x.
- Dag Sehlin; Hillevi Englund; Barbro Simu; Mikael Karlsson; Martin Ingelsson; Fredrik Nikolajeff; Lars Lannfelt; Frida Ekholm Pettersson; Large Aggregates Are the Major Soluble Aβ Species in AD Brain Fractionated with Density Gradient Ultracentrifugation. PLoS ONE 2012, 7, e32014, 10.1371/journal.pone.0032014.


