 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vassilis Souliotis | + 2584 word(s) | 2584 | 2019-12-30 09:03:59 | | | |
| 2 | Vassilis Souliotis | Meta information modification | 2584 | 2020-01-17 19:51:43 | | | | |
| 3 | Vassilis Souliotis | -1126 word(s) | 1458 | 2020-10-30 08:02:43 | | |
Video Upload Options
The DNA damage response and repair (DDR/R) network, a sum of hierarchically structured signaling pathways that recognize and repair DNA damage, and the immune response to endogenous and/or exogenous threats, act synergistically to enhance cellular defence. On the other hand, a deregulated interplay between these systems underlines inflammatory diseases including malignancies and chronic systemic autoimmune diseases, such as systemic lupus erythematosus, systemic sclerosis, and rheumatoid arthritis. Recent data demonstrate accumulation of endogenous DNA damage in peripheral blood mononuclear cells from these patients, which is related to augmented formation of DNA damage and epigenetically regulated functional abnormalities of fundamental DNA repair mechanisms. Since endogenous DNA damage accumulation has serious consequences for cellular health, including genomic instability and enhancement of an aberrant immune response, these results can be exploited for understanding pathogenesis and progression of systemic autoimmune diseases, as well as for the development of new treatments.
Introduction
The DNA damage response and repair (DDR/R) network is a hierarchically structured mechanism, consisting of sensors, mediators, transducers, and effectors, which recognize any defects during the cell cycle and assign the proper repair process[1]. In case of unrepaired lesions and depending on the extent and type of damage, the cell either passes the mutated genome to its offspring or is neutralized by programmed cell death (apoptosis) or senescence[2]. To compensate for the many types of DNA damage that occur, cells have developed six major DNA repair mechanisms wherein each corrects a different subset of lesions: nucleotide excision repair (NER)[3], base excision repair (BER)[4], mismatch repair (MMR)[5], double-strand breaks repair (DSBs/R)[6], interstrand cross-link repair (ICL/R)[7][7], and direct repair pathway[8].
Recently, the interplay between DDR/R and innate immune response has been suggested. Indeed, accumulating data demonstrate that deregulation of DNA repair mechanisms result in the accumulation of cytosolic single- and double-stranded DNAs that can act as potent immunostimulators through the induction of the cGAS-STING-IRF3 pathway and the production of type I interferon[9][10][11][12][13][14]. On the contrary, loss of immune homeostasis and prolonged inflammatory response can lead to DNA damage and activate the DDR/R network, thus indicating a bi-directional relationship between DDR/R and immune response[2][15][16][17][18][19][20][21]. Interestingly, the presence of DSBs per se has been shown to induce type I IFN production. Treatment of healthy donor-derived primary monocytes with etoposide, mitomycin C or adriamycin, three DSB-inducing drugs, was able to induce type I and III IFNs in primary monocytes and various cell lines, suggesting that DDR-induced IFN expression is a universal mechanism that may underline different pathological processes[11]. Furthermore, basic components of DSB repair were shown to be responsible for the production of cytoplasmic ssDNA, which seems to be the main immunostimulant[22].
Important data supporting that defective DNA repair primes innate immune response comes from ataxia-telangiectasia (AT), a neurodegenerative disorder associated with mutations of the central DNA repair kinase ATM[12]. In summary, ATM deficiency leads to the accumulation of DNA damage, exportation of damaged ssDNA and dsDNA into the cytoplasm, activation of the CGAS-STING pathway, and finally type I IFN production that primed cells for response to exogenous or endogenous stimuli such as viral or bacterial infections (Figure 1). Moreover, Günther and colleagues suggested that defective ribonucleotide removal and accumulation of base lesions and low-grade DNA damage “primed” immune response (ImmR)[23].
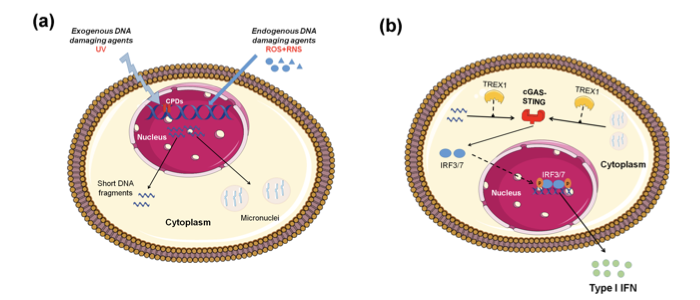
Figure 1. Induction of type I IFN expression by endogenous DNA damage. (A) Exogenous and/or endogenous genotoxic agents may lead to the accumulation of DNA damage in the nucleus, followed by exportation of damaged DNA into the cytoplasm and the induction of micronuclei. (B) Damaged cytoplasmic DNA, if it is not cleared by the exonuclease Trex1, activates the cGAS-STING-IRF3 pathway and the production of type I IFN.
The DDR/R Network in Systemic Autoimmune Diseases
The first hint that abnormalities in DDR/R pathway may be involved in the pathogenesis of autoimmunity comes from the increased frequency of polymorphisms of central molecules involved in the DDR/R pathway such as TREX1[24]. Moreover, autoantibodies against components of the DDR pathway have been detected in approximately 10-20% of patients with systemic lupus erythematosus (SLE)[25]. Some known targets of autoantibodies in SLE are the two subunits of Ku protein (Ku70 and Ku80), DNA ligase IV, XRCC4, DNA-PK, PARP, Mre11 and Werner protein[26][27]. Peripheral blood mononuclear cells (PBMCs) from SLE patients display defects in two main DNA repair pathways, namely, NER and DSB repair. SLE patients with nephritis have approximately 3-5 times higher intrinsic DNA damage compared with healthy controls. Of interest, patients with quiescent disease also exhibited increased levels of DNA damage, although lower than patients with nephritis[10]. Recent studies suggest that either dysregulated apoptosis or defects in dead cell clearance contribute to the perpetuation of autoimmunity and SLE pathogenesis. Studies revealed a significantly higher percentage of apoptotic cells in SLE patients than in controls, which was also positively correlated with the number of plasmacytoid dendritic cells, the major type I IFN-a producer[28]. Interestingly, our previous studies have shown that genotoxic drug-induced apoptosis rates were higher in PBMCs from quiescent SLE patients than healthy controls and correlated inversely with DNA repair efficiency, supporting the hypothesis that accumulation of DNA damage contributes to increased apoptosis[9][10]
Oxidative damage has been implicated in the development and perpetuation of systemic sclerosis (SSc)[29]. Indeed, in fibroblasts isolated from the skin of patients with diffuse SSc, levels of ROS and type I collagen are significantly higher and the amounts of free thiol are significantly lower when compared to normal fibroblasts[30]. Moreover, sera from patients with diffuse SSc and lung fibrosis contain elevated levels of advanced oxidation protein products (AOPPs) compared to sera from healthy individuals or from patients with limited SSc and no lung fibrosis[31]. Furthermore, increased DNA damage levels have also been detected in the peripheral blood of patients with SSc, regardless of disease subtype (diffuse or limited SSc) or treatment[32].
The role of oxidative DNA damage and aberrations of the DDR/R network have been long studied in rheumatoid arthritis (RA)[33]. P53 mutations and overexpression were characteristically detected in the synovium of patients with RA[34]. Immunohistochemical analysis of RA synovial tissues revealed compensatory up-regulation of MMR enzymes, especially in the synovial lining, which, however, did not completely invert the observed oxidative damage[35]. Moreover, increased endogenous DNA damage levels in peripheral blood (PBMCs or granulocytes) are observed in patients with RA compared to healthy controls[36][37].
Conclusion
The DDR/R network and the ImmR act synergistically to ensure genomic stability and cell homeostasis, therefore a balance shift in DDR/R may negatively affect ImmR and the opposite may also occur. Guided by this notion, we propose that epigenetically regulated functional abnormalities of DNA repair mechanisms (i.e. downregulation of DDR/R-related genes and condensed chromatin structure that result in defective repair) and increased endogenous DNA damage formation, partly due to the induction of oxidative stress, may result in the augmented accumulation of DNA damage (both SSBs and DSBs) in patients with systemic autoimmune diseases (Figure 2). This accumulation may trigger the induction of apoptosis, which facilitates autoantibody production, as well as the generation of damaged cytosolic DNA and micronuclei that both can act as potent immunostimulators through the induction of the type I IFN pathway, leading to systemic autoimmune disease expression. Notably, some of the components cGAS-STING-IRF3 pathway and the production of type I IFN, leading to systemic autoimmune disease are partially reversible following histone hyperacetylation by HDAC inhibitors.
Recent data have shown that treatment of human SLE-derived PBMCs with the HDACi vorinostat results in hyperacetylation of histone H4, chromatin decondensation, restoration of the DNA repair capacity, and decreased apoptosis rates[10]. These results are in line with previous data, showing that HDACi ameliorate disease in lupus mouse models[38][39][40], and that treatment of lupus-prone Mrl/lpr mice with the HDACi panobinostat significantly reduced circulating naïve B and plasma cell numbers and the levels of autoantibodies[41]. More importantly, in children with systemic-onset juvenile idiopathic arthritis, the HDACi givinostat was found to be safe and beneficial, particularly in reducing the arthritic features, suggesting that HDACi may have important clinical applications in the treatment of systemic autoimmunity[42].
Taken together, the results reviewed herein suggest that the deregulated interplay between DDR/R and ImmR plays a crucial role in the pathogenesis and progression of systemic autoimmune diseases. Thus, unraveling the molecular mechanisms of this interplay can be exploited for understanding pathogenesis and progression of these diseases, as well as to discover new treatment opportunities in the field.
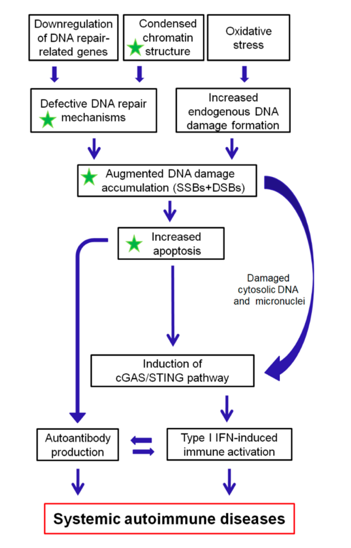
Figure 2. A proposed model of systemic autoimmune disease promotion by epigenetically regulated functional abnormalities of the DNA damage response and repair (DDR/R) network and oxidative stress. The green asterisk denotes partial reversibility following histone hyperacetylation. SSBs: single-strand breaks, DSBs: double-strand breaks.
References
- Ioannis S. Pateras; Sophia Havaki; Xenia Nikitopoulou; Konstantinos Vougas; Paul A. Townsend; Michalis I. Panayiotidis; Alexandros G. Georgakilas; Vassilis G. Gorgoulis; The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol Ther. 2015, 154, 36-56, 10.1016/j.pharmthera.2015.06.011.
- Stephen Philip Jackson; Jiri Bartek; The DNA-damage response in human biology and disease. Nature 2009, 461, 1071-8, 10.1038/nature08467.
- Sarah C. Shuck; Emily A. Short; John J. Turchi; Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res 2008, 18, 64-72, 10.1038/cr.2008.2.
- Dmitry O. Zharkov.; Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565, 10.1007/s00018-008-7543-2.
- Sarah A. Martin; Christopher J. Lord; Alan Ashworth; Therapeutic Targeting of the DNA Mismatch Repair Pathway. Clin. Cancer Res. 2010, 16, 5107-5113, 10.1158/1078-0432.ccr-10-0821.
- Alberto Ciccia; Stephen J. Elledge; The DNA Damage Response: Making it safe to play with knives. Mol Cell. 2010, 40, 179-204, 10.1016/j.molcel.2010.09.019.
- Andrew J. Deans; Stephen C. West; DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011, 11, 467-80, 10.1038/nrc3088.
- Birgitta I Hiddinga; Pauwels, P.; Janssens, A.; van Meerbeeck, J.P. O6-Methylguanine-DNA methyltransferase (MGMT): A drugable target in lung cancer? Lung Cancer (Amst. Neth.) 2017, 107, 91–99, 10.1016/j.lungcan.2016.07.014.
- Vassilis L. Souliotis; Petros P. Sfikakis; Increased DNA double-strand breaks and enhanced apoptosis in patients with lupus nephritis, Lupus 2014, 24, 804-815, 10.1177/0961203314565413.
- Vassilis L. Souliotis; Konstantinos Vougas; Vassilis G. Gorgoulis; Petros P. Sfikakis; Defective DNA repair and chromatin organization in patients with quiescent systemic lupus erythematosus, Arthritis Res Ther. 2016, 18, 182, 10.1186/s13075-016-1081-3.
- Brzostek-Racine, S.; Gordon, C.; Van Scoy, S.; Reich, N.C; The DNA damage response induces IFN, J. Immunol. 2011, 187, 5336–5345, 10.4049/jimmunol.1100040.
- Anetta Hartlova; Saskia F. Erttmann; Faizal Am. Raffi; Anja M. Schmalz; Ulrike Resch; Sharath Anugula; Stefan Lienenklaus; Lisa M. Nilsson; Andrea Kröger; Jonas A. Nilsson; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity, Immunity 2015, 42, 332-343, 10.1016/j.immuni.2015.01.012.
- Lorenzo Galluzzi; Aitziber Buqué; Oliver Kepp; Laurence Zitvogel; Guido Kroemer; Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents, Cancer Cell 2015, 28, 690-714, 10.1016/j.ccell.2015.10.012.
- Yu J. Shen; Nina Le Bert; Anuja A. Chitre; Christine Xing’Er Koo; Xing H. Nga; Samantha S.W. Ho; Muznah Khatoo; Nikki Y. Tan; Ken J. Ishii; Stephan Gasser; et al. Genome-Derived Cytosolic DNA Mediates Type I Interferon-Dependent Rejection of B Cell Lymphoma Cells; Cell Rep. 2015, 11, 460-473, 10.1016/j.celrep.2015.03.041.
- Lisiane B. Meira; James M. Bugni; Stephanie L. Green; Chung-Wei Lee; Bo Pang; Diana Borenshtein; Barry H. Rickman; Arlin B. Rogers; Catherine A. Moroski-Erkul; Jose L. McFaline; et al.David B. SchauerPeter C. DedonJames G. FoxLeona D. Samson; DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice, Journal of Clinical Investigation 2008, 118, 2516-2525, 10.1172/JCI35073.
- Shiho Ohnishi; Ning Ma; Raynoo Thanan; Somchai Pinlaor; Olfat Hammam; Mariko Murata; Shosuke Kawanishi; DNA Damage in Inflammation-Related Carcinogenesis and Cancer Stem Cells, Oxid Med Cell Longev. 2013, 2013, 1-9, 10.1155/2013/387014.
- Timea Palmai-Pallag; Csanad Z. Bachrati; Inflammation-induced DNA damage and damage-induced inflammation: a vicious cycle, Microbes Infect. 2014, 16, 822-832, 10.1016/j.micinf.2014.10.001.
- Dawit Kidane; Wook Jin Chae; Jennifer Czochor; Kristin A. Eckert; Peter M. Glazer; Alfred L. M. Bothwell; Joann B. Sweasy; Interplay between DNA repair and inflammation, and the link to cancer, Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116-39, 10.3109/10409238.2013.875514.
- Fabrícia Lima Fontes; Daniele Maria Lopes Pinheiro; Ana Helena Sales De Oliveira; Rayssa Karla De Medeiros Oliveira; Tirzah Braz Petta Lajus; Lucymara Fassarella Agnez-Lima; Role of DNA repair in host immune response and inflammation, Mutat Res Rev Mutat. Res. 2015, 763, 246-257, 10.1016/j.mrrev.2014.11.004.
- Orsolya Kiraly; Gong, G.; Olipitz, W.; Muthupalani, S.; Engelward, B.P.; Inflammation-Induced Cell Proliferatio Potentiates DNA Damage-Induced Mutations In Vivo, PLoS Genet. 2015, 11, e1004901, 10.1371/journal.pgen.1004901.
- Ana Neves-Costa; Luis F. Moita; Modulation of inflammation and disease tolerance by DNA damage response pathways, FEBS J. 2016, 284, 680-698, 10.1111/febs.13910.
- Erkin Erdal; Syed Haider; Jan Rehwinkel; Adrian L. Harris; Peter J. McHugh; A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1, Genes Dev, 2017, 31, 353-369, 10.1101/gad.289769.116.
- Claudia Günther; Barbara Kind; Martin A.M. Reijns; Nicole Berndt; Manuel Martínez-Bueno; Christine Wolf; Victoria Tüngler; Osvaldo Chara; Young Ae Lee; Norbert Hubner; et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity, J Clin Invest. 2014, 125, 413-424, 10.1172/JCI78001.
- Jessica L. Grieves; Jason M. Fye; Scott Harvey; Jason M. Grayson; Thomas Hollis; Fred W. Perrino; Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease, Proc. Natl. Acad. Sci. USA 2015, 112, 5117–5122, 10.1073/pnas.1423804112.
- Philip W Noble; Bernatsky,S.;Clarke,A.E.;Isenberg,D.A.;Ramsey-Goldman,R.;Hansen,J.E.; DNA-damaging autoantibodies and cancer: The lupus butterfly theory, Nat. Rev. Rheumatol. 2016, 12, 429–434, 10.1038/nrrheum.2016.23.
- Victoria L. Fell; Caroline Schild-Poulter; The Ku heterodimer: Function in DNA repair and beyond, Mutat Res Rev Mutat Res 2015, 763, 15-29, 10.1016/j.mrrev.2014.06.002.
- Hui Luo; Wang, L.; Bao, D.; Wang, L.; Zhao, H.; Lian, Y.; Yan, M.; Mohan, C.; Li, Q.Z.; Novel Autoantibodies Related to Cell Death and DNA Repair Pathways in Systemic Lupus Erythematosus; Genom. Proteom. Bioinform. 2019, 17, 248–259, 10.1016/j.gpb.2018.11.004.
- Ji Won Park; Moon, S.Y.; Lee, J.H.; Park, J.K.; Lee, D.S.; Jung, K.C.; Song, Y.W.; Lee, E.B.; Bone marrow analysis of immune cells and apoptosis in patients with systemic lupus erythematosus, Lupus 2014, 23, 975–985, 10.1177/0961203314531634.
- Paola Sambo; Silvia Svegliati Baroni; Michele Luchetti; Paolo Paroncini; Stefano Dusi; Guido Orlandini; Armando Gabrielli; Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive upregulation of reactive oxygen species generation through the NADPH oxidase complex pathway; Arthritis Rheum. 2001, 44, 2653-2664, 10.1002/1529-0131(200111)44:11<2653::aid-art445>3.0.co;2-1.
- Pei-Suen Tsou; Talia,N.N.; Pinney,A.J.; Kendzicky,A.; Piera Velazquez,S.; Jimenez,S.A.; Seibold,J.R.; Phillips,K.; Koch, A.E.; Effect of oxidative stress on protein tyrosine phosphatase 1B in scleroderma dermal fibroblasts; Arthritis Rheum. 2012, 64, 1978–1989, 10.1002/art.34336.
- Amélie Servettaz; Claire Goulvestre; Niloufar Kavian; Carole Nicco; Philippe Guilpain; Christiane Chéreau; Vincent Vuiblet; Loïc Guillevin; Luc Mouthon; Bernard Weill; et al.Frederic Batteux Selective Oxidation of DNA Topoisomerase 1 Induces Systemic Sclerosis in the Mouse; J. Immunol. 2009, 182, 5855-5864, 10.4049/jimmunol.0803705.
- Gustavo Martelli Palomino; Bassi, C.L.; Wastowski, I.J.; Xavier, D.J.; Lucisano-Valim, Y.M.; Crispim, J.C.O.; Rassi, D.M.; Marques-Netom, J.F.; Sakamoto-Hojo, E.T.; Moreau, P.; et al. Patients with systemic sclerosis present increased DNA damage differentially associated with DNA repair gene polymorphisms, J. Rheumatol. 2014, 41, 458–465, 10.3899/jrheum.130376.
- Lan Shao; DNA Damage Response Signals Transduce Stress From Rheumatoid Arthritis Risk Factors Into T Cell Dysfunction, Front. Immunol. 2018, 9, 3055, 10.3389/fimmu.2018.03055.
- Gary S. Firestein; Echeverri,F.; Yeo,M.; Zvaifler,N.J.; Green,D.R.; Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium, Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900, 10.1073/pnas.94.20.10895.
- Egle Šimelyte; D L Boyle; G S Firestein; DNA mismatch repair enzyme expression, Rheumatic Ann Rheum Dis. 2004, 63, 1695-1699, 10.1136/ard.2003.017210.
- Vassilis L. Souliotis; Nikolaos I. Vlachogiannis; Maria Pappa; Alexandra Argyriou; Petros P. Sfikakis; DNA damage accumulation, defective chromatin organization and deficient DNA repair capacity in patients with rheumatoid arthritis, Clin. Immunol. 2019, 203, 28-36, 10.1016/j.clim.2019.03.009.
- Martelli-Palomino,G.; Paoliello Paschoalato,A.B.; Crispim,J.C.; Rassi,D.M.; Oliveira,R.D.; Louzada,P.; Lucisano-Valim, Y.M.; Donadi, E.A.; DNA damage increase in peripheral neutrophils from patients with rheumatoid arthritis is associated with the disease activity and the presence of shared epitope, Clin. Exp. Rheumatol. 2017, 35, 247–254, PMID: 27908303.
- Nilamadhab Mishra; Christopher M. Reilly; Doris R. Brown; Phil Ruiz; Gary S. Gilkeson; Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse, J Clin Invest. 2003, 111, 539-552, 10.1172/JCI200316153.
- Nicole L. Regna; Miranda D. Vieson; Xin M. Luo; Cristen B. Chafin; Abdul Gafoor Puthiyaveetil; Sarah E. Hammond; David L. Caudell; Matthew B. Jarpe; Christopher M. Reilly; Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice, Clin Immunol. 2016, 162, 58-73, 10.1016/j.clim.2015.11.007.
- Jilian Ren; X. Liao; M. D. Vieson; M. Chen; R. Scott; J. Kazmierczak; X. M. Luo; C. M. Reilly; Selective HDAC6 inhibition decreases early stage of lupus nephritis by down‐regulating both innate and adaptive immune responses, Clin Exp Immunol. 2017, 191, 19-31, 10.1111/cei.13046.
- Michaela Waibel; Ailsa J Christiansen; Margaret L Hibbs; Jake Shortt; Sarah A Jones; Ian Simpson; Amanda Light; Kristy O'Donnell; Eric F Morand; David M Tarlinton; Ricky W Johnstone; Edwin D Hawkins; Manipulation of B-cell responses with histone deacetylase inhibitors, Nat Commun. 2015, 6, 6838, 10.1038/ncomms7838.
- Jelena Vojinovic; Nemanja Damjanov; Carmine D'urzo; Antonio Furlan; Gordana Susic; Srdjan Pasic; Nicola Iagaru; Mariana Stefan; Charles A. Dinarello; Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis, Arthritis Rheum. 2011, 63, 1452-1458, 10.1002/art.30238.


