 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel Foulkes | + 2299 word(s) | 2299 | 2019-12-26 05:04:21 | | | |
| 2 | Nicole Yin | -573 word(s) | 1726 | 2020-01-15 09:50:36 | | | | |
| 3 | Nicole Yin | Meta information modification | 1726 | 2020-07-31 08:02:02 | | | | |
| 4 | Nicole Yin | -12 word(s) | 1714 | 2020-10-23 03:54:19 | | |
Video Upload Options
The opportunistic pathogen Pseudomonas aeruginosa employs the type III secretion system (T3SS) and four effector proteins, ExoS, ExoT, ExoU, and ExoY, to disrupt cellular physiology and subvert the host's innate immune response. Of the effector proteins delivered by the T3SS, ExoU is the most toxic. In P. aeruginosa infections, where the ExoU gene is expressed, disease severity is increased with poorer prognoses. This is considered to be due to the rapid and irreversible damage exerted by the phospholipase activity of ExoU, which cannot be halted before conventional antibiotics can successfully eliminate the pathogen. Here we explore what is currently known about ExoU and its potential as a therapeutic target, highlighting some of the small molecule ExoU inhibitors that have been discovered from screening approaches.
1. Introduction
Although the mechanisms of ExoU regulation are not fully understood, evidence for ubiquitin-binding as an activator and recruitment to the plasma membrane and oligomerisation followed by phospholipase A2 activity have been described in ExoU associated pathology[1][2]. The interactions of certain eukaryotic co-factors with ExoU results in multiple dynamic conformational changes that could potentially be targeted by small molecules to attenuate ExoU activity in clinical infections (Figure 1)[3][4][5][6].
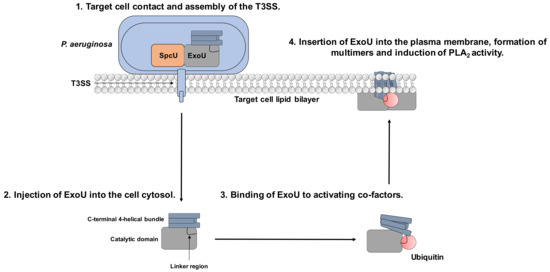
Figure 1. Mechanism of ExoU toxicity and potential for targeting with small molecules. 1. P. aeruginosa binds to the target cell, which stimulates T3SS assembly. 2. ExoU is injected to the host cell via the T3SS. 3. Once in the cellular cytoplasm, it interacts with the eukaryotic host co-factor ubiquitin. 4. ExoU localises to the plasma membrane and oligomerises to stimulate full catalytic activity, leading to cellular lysis. The linker region links the catalytic domain to the 4-helical membrane localisation domain (MLD) and is the proposed binding site for ubiquitin. The 4-helical MLD domain is in the C-terminus of the protein and is responsible for interaction with PIP2 and insertion into the host cell plasma membrane. Compounds that could potentially subvert ExoU mediated cytotoxicity could: prevent ExoU secretion from P. aeruginosa, bind to the substrate-binding site in the catalytic domain, prevent ubiquitin-binding, or inhibit multimerisation and association with PIP2.
In 2003, methyl arachidonyl fluorophosphonate (MAFP), an irreversible active site-directed promiscuous phospholipase inhibitor, was shown to protect Chinese Hamster Ovary (CHO) cells from ExoU mediated cell lysis after infection with the PA103 clinical isolate strain of P. aeruginosa[1] . The arachidonic acid analogue MAFP is a pan PLA2 inhibitor (Figure 2), which possess a phosphate group that covalently binds to the phospholipase catalytic serine residue. Although high concentrations of MAFP (67.5 μM) were required to achieve measurable inhibition of CHO cell lysis, this was the first empirical proof that ExoU catalytic activity could be targeted by a small molecule inhibitor. This also validated the assertion that clinical phospholipase inhibitors, developed to target endogenous human phospholipases across a broad spectrum of diseases[7] could potentially have ExoU inhibiting activity. Such compounds could be repurposed to circumvent the pathological consequences of ExoU expression[8].
A subsequent study employed a high-throughput screen to discover compounds that could inhibit T3SS mediated cytotoxicity, with ExoU as the only cytotoxic effector[6] . By maintaining CHO cell viability in the presence of ExoU expressing strains of P. aeruginosa, 9H-fluorene-4-carboxamide, the designated pseudolipasin A (Figure 2), was identified as a promising compound lead. Further experimentation confirmed that pseudolipasin A did not inhibit type III secretion or type III injection of ExoU into CHO cells, but rather inhibited ExoU downstream of injection by the T3SS[6]. Relatively high concentrations of pseudolipasin A (5–10 µM) were required to subvert the lytic effects of ExoU after CHO cells were infected with P. aeruginosa. This was in agreement with a 7 μM reported IC50 value for inhibition for ExoU catalytic activity in vitro, which was determined by a phospholipase assay using recombinant ExoU expressed in E. coli.
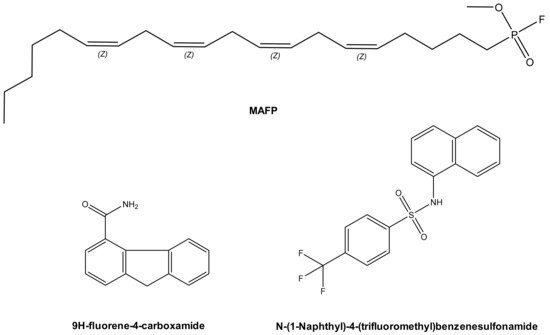
Figure 2. Prospective ExoU small molecule inhibitors. MAFP (Methoxy arachidonyl fluorophosphonate) is a non-specific PLA2 inhibitor, which is an analogue of arachidonic acid. Chemical structures of compounds previously distinguished to inhibit ExoU from two independent screens are also illustrated.
In an independent screen, an arylsulphonamide was found to be effective at reducing the cytotoxic effect exerted by induced recombinant ExoU expression in yeast[9]. Saccharomyces cerevisiae were transformed with pDH105, which encoded ExoU cDNA. Upon induction with copper, ExoU expression led to S. cerevisiae lysis except in the presence of 5 µM of a ‘hit’ arylsulfonamide compound (Figure 2)[9]. Synthesis of a small series of arylsulfonamides, comprising the N-(naphthalen-1-yl)benzenesulfonamide core, validated that this scaffold, when coupled to various commercially available sulfonyl chlorides, yielded compounds with differential abilities to mitigate ExoU mediated cell death[9]. Even though none were as effective as pseudolipasin A, the finding highlighted a novel pharmacological scaffold that could potentially be developed as an inhibitor of ExoU activity. A full biochemical exploration of the inhibitory mechanisms of these compounds remains to be undertaken, and it is not known whether or not these arylsulphonamides inhibit ExoU phospholipase activity in vitro.
Aside from modulating catalytic activity by binding to the ExoU active site, alternate modes of inhibition could occur. For instance, compound binding may inhibit ubiquitin-binding, conformational rearrangement, oligomerization, or phospholipid binding (Figure 1). It also remains to be tested whether or not the arylsulphonamide compounds can effectively protect mammalian cells after P. aeruginosa infection in vitro[9].
In order to capitalise on pseudolipasin A and certain arylsulphonamides as potential drug leads, further avenues of structure–activity relationships require exploration. As well as understanding mechanisms and conformations relevant to compound binding, structure-guided drug design could be used to modify pseudolipasin A[6] and the arylsulphonamides[9] to generate selective and more potent ExoU inhibitors. Analogues of the former compound, with a fluorene scaffold, appear to have cytotoxic effects on mammalian cells[9], which could preclude further development of pseudolipasin A as an ExoU inhibitor. Prospectively, the arylsulfonamide potential ExoU inhibitors display limited cytotoxicity in HEK293T cells[9]. Crystal structures of ExoU in complex with ubiquitin, phospholipid and/or compounds would be valuable for informing structure-guided design of such compounds. ExoU appears to be a dynamic protein, and the study of such conformations associated with cofactor and ligand binding might be explored by cryogenic electron microscopy (cryo EM). As ExoU is catalytically inactive when bound to its cognate chaperone SpcU[10] , the already available co-crystal structures of ExoU and SpcU (PDB entries 3TU3 and 4AKX)[2][11] could potentially serve as a foundation for structure-based design of inhibitors or peptides that mimic the interface between the N-terminal domain of ExoU and SpcU. Perhaps the structures of ExoU and SpcU could be explored by in silico-based screening approaches in order to discover novel potential ExoU ligands.
2. Allosteric Inhibitors
In order to find novel ExoU inhibitors, multiple screening strategies could be explored to evaluate compounds’ ability to inhibit phospholipase A2 ExoU activity, translocation through the T3SS, and interaction with host co-factors. Allosteric inhibitors of ExoU may offer distinct efficacious and selective advantages over small molecules occupying the phospholipase substrate-binding pocket. Molecules that bind to conserved regions of the active site, encompassing the Serine-Aspartic acid catalytic dyad, may have limited selectivity and produced unwanted ‘off-target’ cytotoxic effects. Indeed, pseudolipasin A does not inhibit any of human group IID, IIE, V, X, or XII PLA2 phospholipases tested[6]. Allosteric inhibitors exert their effects either by indirectly targeting catalytic activity or by modulating non-catalytic function, such as interactions with activating co-factors. They generally achieve better target precision through binding to specific residues, outside of the conserved active site. Such approaches have become popular for pharmacological targeting of disease-associated protein kinases[12]. Exploiting the ubiquitin-binding site of ExoU, which has been modelled in silico with the use of biophysical analysis and available crystallographic evidence[3], could serve as a point of allosteric inhibition that is selective and potent to ExoU activity inhibition. Such inhibitors could offer the advantage of being non-substrate competitive and, therefore, potentially, achieve superior potency. To this end, screening approaches to interrogate ExoU interaction with ubiquitin and PIP2 in the presence of prospective compounds would need to be developed.
An example of allosteric targeting of phospholipases is compound VU0364739. This is a phospholipase D2 (PLD2)-selective inhibitor, which occupies an allosteric site and blocks PIP2 binding[13]. The selectivity of this compound was evidenced by a starkly increased IC50 value for inhibition of the closely related PLD1 isoform of 1500 nM compared to the IC50 value for PLD2 of 20 nM. VU0364739 is structurally similar to previously reported piperidine benzimidazolone-based allosteric inhibitors of AKT[14]. The PIP2-binding site forms a hydrophobic pocket in the pleckstrin homology (PH) domain of PLD2, and the mode of inhibition is proposed to be akin to that facilitated by allosteric AKT compounds[13][15]. Perhaps a similar allosteric mechanism of inhibition of ExoU could be discovered, whereby certain classes of compound occupy a site within the C-terminal domain of ExoU in order to mitigate interaction with PIP2.
3. Ligand Repurposing Approaches
The activities of phospholipase A2 enzymes is a contributing factor to various inflammatory pathological conditions, including arthritis, cardiovascular, and autoimmune diseases[7]. Modulation of their catalytic activity has become of major interest to pharmaceutical companies in recent years for the development of novel therapeutics[7]. De novo drug discovery, usually performed in the pharmaceutical industry, is an expensive and long process. Drug repurposing approaches have been used across a broad spectrum of diseases[16], where many “repurposed” drugs, such as antibiotics, antivirals, and kinase inhibitors, have had clinical success in treatment areas beyond their originally intended use. In a recent study, certain kinase inhibitors, including an inhibitor of PDK1, were able to function as an adjuvant in colistin-resistant bacteria, potentiating the antimicrobial effect in Gram-negative and Gram-positive bacteria[17]. This demonstrates that cross-reactivity of inhibitors originally intended to target eukaryotic proteins may be extended to target prokaryotic virulence effectors. Repurposing of clinical eukaryotic phospholipase inhibitors, to instead target the phospholipase A2 activity of ExoU, may have the advantage of entering the clinic more quickly, as they have already been tested for safety in human beings[8].
There may yet exist phospholipase A2 inhibitors that are cross-reactive with ExoU, which could be utilised when ExoU expression by P. aeruginosa is a driver of disease progression. Compounds exhibiting such cross-reactivity with ExoU could potentially serve as a foundation for the development of more selective ExoU inhibitors. Medicinal chemistry approaches, including in silico modelling, organic synthesis, and the study of structure-activity relationships may provide new chemicals as potential novel anti-ExoU pharmaceutical agents.
References
- Rebecca M. Phillips; David A. Six; Edward A. Dennis; Partho Ghosh; Kazu Matsumoto; Masafumi Shionyu; Mitiko Go; Katsuji Shimizu; Tamayuki Shinomura; Koji Kimata; Hideto Watanabe; In Vivo Phospholipase Activity of the Pseudomonas aeruginosa Cytotoxin ExoU and Protection of Mammalian Cells with Phospholipase A2 Inhibitors. Journal of Biological Chemistry 2003, 278, 41326-41332, 10.1074/jbc.m302472200.
- Andrei S. Halavaty; Minika Borek; Gregory H. Tyson; Jeff L. Veesenmeyer; Ludmilla Shuvalova; George Minasov; Zbyszek Otwinowski; Alan R. Hauser; Wayne F. Anderson; Structure of the Type III Secretion Effector Protein ExoU in Complex with Its Chaperone SpcU. PLOS ONE 2012, 7, e49388, 10.1371/journal.pone.0049388.
- Maxx H. Tessmer; David M. Anderson; Adam M. Pickrum; Molly O. Riegert; Rocco Moretti; Jens Meiler; Jimmy B. Feix; Dara W. Frank; Identification of a ubiquitin-binding interface using Rosetta and DEER. Proceedings of the National Academy of Sciences 2018, 115, 525-530, 10.1073/pnas.1716861115.
- Gregory H. Tyson; Andrei S. Halavaty; Hyunjin Kim; Brett Geissler; Mallory Agard; Karla J. Satchell; Wonhwa Cho; Wayne F. Anderson; Alan R. Hauser; A Novel Phosphatidylinositol 4,5-Bisphosphate Binding Domain Mediates Plasma Membrane Localization of ExoU and Other Patatin-like Phospholipases*. Journal of Biological Chemistry 2014, 290, 2919-2937, 10.1074/jbc.M114.611251.
- Angelica Zhang; Jeffrey L. Veesenmeyer; Alan R. Hauser; Phosphatidylinositol 4,5-Bisphosphate-Dependent Oligomerization of the Pseudomonas aeruginosa Cytotoxin ExoU. Infection and Immunity 2017, 86, e00402-17, 10.1128/iai.00402-17.
- Vincent T. Lee; Stefan Pukatzki; Hiromi Sato; Eriya Kikawada; Anastasia A. Kazimirova; Jin Huang; Xiaohua Li; Jonathan P. Arm; Dara W. Frank; Stephen Lory; Pseudolipasin A Is a Specific Inhibitor for Phospholipase A2 Activity of Pseudomonas aeruginosa Cytotoxin ExoU▿ †. Infection and Immunity 2006, 75, 1089-1098, 10.1128/IAI.01184-06.
- Aikaterini Nikolaou; Maroula G. Kokotou; Sofia Vasilakaki; George Kokotos; Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2.. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2018, 1864, 941-956, 10.1016/j.bbalip.2018.08.009.
- Ju Bao; Bindumadhav Marathe; Elena A. Govorkova; Jie J. Zheng; Drug Repurposing Identifies Inhibitors of Oseltamivir-Resistant Influenza Viruses.. Angewandte Chemie International Edition 2016, 55, 3438-41, 10.1002/anie.201511361.
- Doran Kim; Jihae Baek; Jiho Song; Hyeyoung Byeon; Hyeyoung Min; Kyung Hoon Min; Identification of arylsulfonamides as ExoU inhibitors. Bioorganic & Medicinal Chemistry Letters 2014, 24, 3823-3825, 10.1016/j.bmcl.2014.06.064.
- Andrei S. Halavaty; Minika Borek; Gregory H. Tyson; Jeff L. Veesenmeyer; Ludmilla Shuvalova; George Minasov; Zbyszek Otwinowski; Alan R. Hauser; Wayne F. Anderson; Structure of the Type III Secretion Effector Protein ExoU in Complex with Its Chaperone SpcU. PLOS ONE 2012, 7, e49388, 10.1371/journal.pone.0049388.
- Claire Gendrin; Carlos Contreras-Martel; Stéphanie Bouillot; Sylvie Elsen; David Lemaire; Dimitrios A. Skoufias; Philippe Huber; Ina Attrée; Andréa Dessen; Structural Basis of Cytotoxicity Mediated by the Type III Secretion Toxin ExoU from Pseudomonas aeruginosa. PLOS Pathogens 2012, 8, e1002637, 10.1371/journal.ppat.1002637.
- Alejandro E. Leroux; Jörg O. Schulze; Ricardo M. Biondi; AGC kinases, mechanisms of regulation and innovative drug development. Seminars in Cancer Biology 2018, 48, 1-17, 10.1016/j.semcancer.2017.05.011.
- H. Alex Brown; Paul G. Thomas; Craig W. Lindsley; Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nature Reviews Drug Discovery 2017, 16, 351-367, 10.1038/nrd.2016.252.
- Craig W. Lindsley; Zhijian Zhao; William H. Leister; Ronald G. Robinson; Stanley F. Barnett; Deborah Defeo-Jones; Raymond E. Jones; George D. Hartman; Joel R. Huff; Hans E. Huber; et al.Mark E. Duggan Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorganic & Medicinal Chemistry Letters 2005, 15, 761-764, 10.1016/j.bmcl.2004.11.011.
- Stanley F. Barnett; Deborah Defeo-Jones; Sheng Fu; Paula J. Hancock; Kathleen M. Haskell; Raymond E. Jones; Jason A. Kahana; Astrid M. Kral; Karen Leander; Ling L. Lee; et al.John MalinowskiElizabeth M. McAvoyDebbie D. NahasRonald G. RobinsonHans E. Huber Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochemical Journal 2005, 385, 399-408, 10.1042/bj20041140.
- 16. Schein, C.H. Repurposing approved drugs on the pathway to novel therapies. Med. Res. Rev. 2019, 10.1002/med.21627.
- William T. Barker; Ansley M. Nemeth; Sara M. Brackett; Akash K. Basak; Courtney E. Chandler; Leigh A. Jania; William J. Zuercher; Roberta J. Melander; Beverly H. Koller; Robert K. Ernst; et al.Christian Melander Repurposing Eukaryotic Kinase Inhibitors as Colistin Adjuvants in Gram-Negative Bacteria.. ACS Infectious Diseases 2019, 5, 1764-1771, 10.1021/acsinfecdis.9b00212.


