 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anthony Maxwell | + 1587 word(s) | 1587 | 2020-12-11 04:57:46 | | | |
| 2 | Vicky Zhou | Meta information modification | 1587 | 2020-12-23 08:28:55 | | | | |
| 3 | Vicky Zhou | Meta information modification | 1587 | 2020-12-23 08:44:30 | | | | |
| 4 | Vicky Zhou | Meta information modification | 1587 | 2020-12-25 01:52:37 | | |
Video Upload Options
Fluoroquinolones (FQs) are arguably among the most successful antibiotics of recent times. They have enjoyed over 30 years of clinical usage and become essential tools in the armoury of clinical treatments. FQs target the bacterial enzymes DNA gyrase and DNA topoisomerase IV, where they stabilise a covalent enzyme-DNA complex in which the DNA is cleaved in both strands. This leads to cell death and turns out to be a very effective way of killing bacteria. However, resistance to FQs is increasingly problematic, and alternative compounds are urgently needed.
1. Introduction
The quinolone antibiotics (Figure 1) are the most successful class of topoisomerase inhibitors to date. They are synthetic antimicrobials with the initial compound, nalidixic acid, being discovered as a by-product of chloroquine synthesis in 1962[1][2]. They are used to treat bacterial infections caused by both Gram-positive and Gram-negative bacteria, including, but not limited to, urinary tract infections (UTIs), pyelonephritis, gastroenteritis, sexually-transmitted diseases, such as Gonorrhoea, tuberculosis[3], prostatitis, community-acquired pneumonia and skin and soft-tissue infections[4][5]. However, due to an increase in resistance and issues surrounding toxicity, their use in the treatment of mild infections has been contraindicated[6]. The global rise in antibiotic resistance has galvanised research into new antibiotics against both well-established targets and new targets. It has also sparked further research into antibiotics whose mode of killing is less well-established, as well as how bacteria become resistant to them. This is certainly true in the case of quinolones.
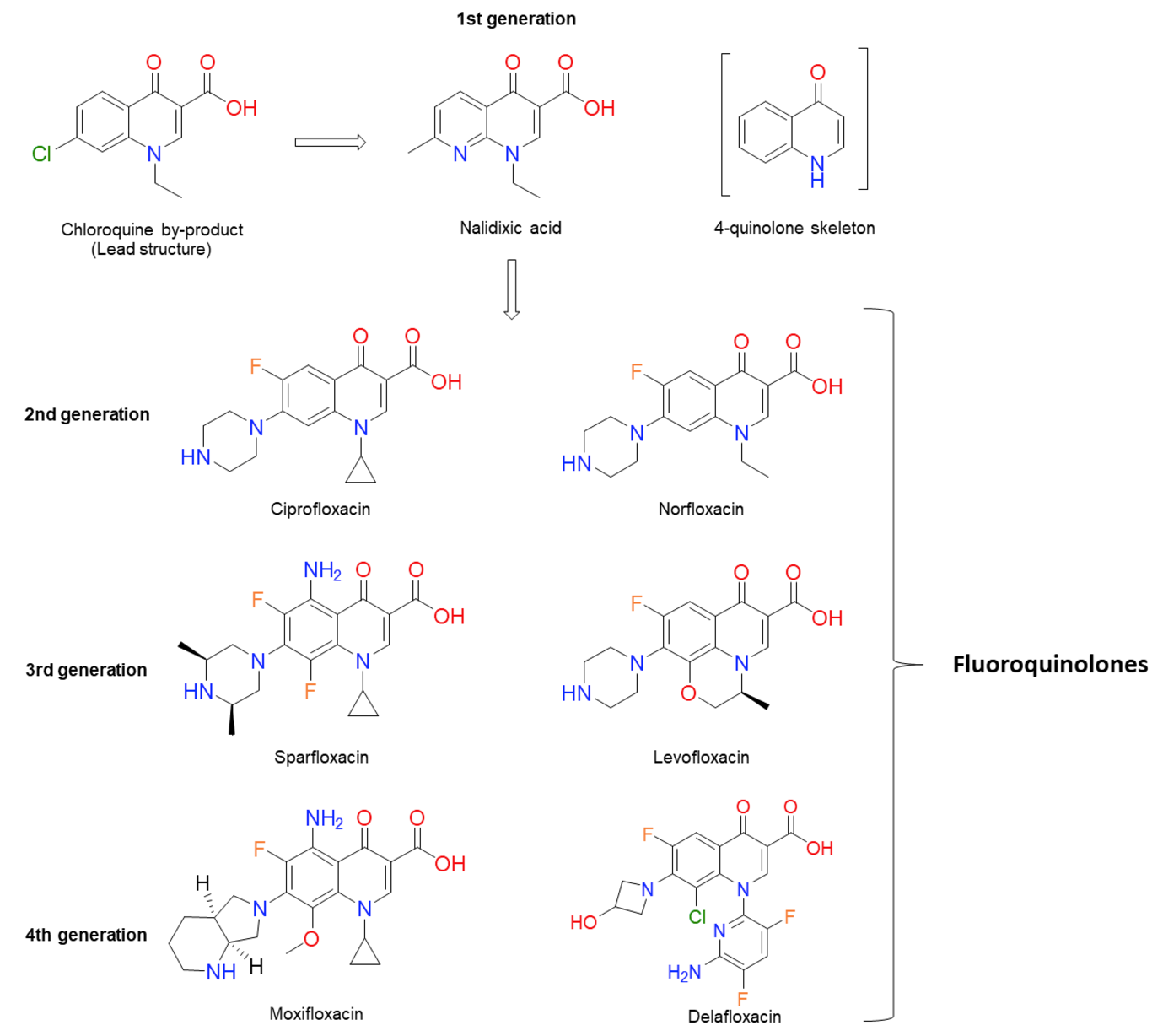
Figure 1. Chemical structures of several significant fluoroquinolones. Nalidixic acid (the first “quinolone”) is shown, along with the chloroquine by-product inspiring its synthesis. Note that nalidixic acid lacks the 4-quinolone core and instead contains a 1,8-naphthyridine nucleus.
2. Quinolone-Induced AMR
Along with the increase in FQ resistance that is seen with FQ use, there is also evidence that FQs may increase resistance to non-FQ antibiotics[7][8][9][10][11], particularly under sublethal or sub-MIC exposure. Treatments with sublethal FQs have been shown to increase mutation, recombination and persister formation rates, often leading to an increase in the frequency of resistance to non-quinolone antibiotics.[12][13][14][15][16][17][18]
2.1. Treatment with Quinolones Increases Resistance to Non-Quinolone Antibiotics (QIAR)
There is growing evidence, particularly from the livestock and veterinary sectors, that FQ use can lead to an increase in antibiotic-resistant isolates[19]. This resistance can be FQ resistance, resistance to non-FQ antimicrobials or multidrug resistance. Pereira et al.[9] looked at the resistance profiles of E. coli isolates from pre-weaned calves that were treated with enrofloxacin or the cephalosporin ceftiofur for diarrhoea and respiratory diseases. They found that 77% of the isolates from the FQ-treated calves showed resistance to three or more antimicrobials, including ciprofloxacin, streptomycin, tetracycline, ampicillin, ceftiofur and chloramphenicol. In the study, only the calves treated with the FQ were significantly more likely to have non-susceptible E. coli isolates[9]. Similarly, a study on healthy chickens found an increase in the number of isolates resistant to doxycycline, amoxicillin and enrofloxacin in the commensal E. coli populations after treatment with enrofloxacin[7]. This was mirrored in more recent studies on commensal E. coli isolates from chickens and turkeys that were treated with enrofloxacin [10][11]. In the former study, multidrug resistance was identified in E. coli isolates from chickens treated with the FQ for Salmonella sp. infections[11], whilst, in the latter, E. coli isolates from turkeys treated with enrofloxacin were found to be resistant to ampicillin, despite ampicillin not being used in the study[10]. Further evidence comes from Sa. Typhimurium clinical isolates from pigs, which showed multidrug resistance following a single treatment of marbofloxacin below the mutant prevention concentration[20]. This phenomenon is not peculiar to isolates from livestock; a strain of multidrug resistant Sa. enteritidis was isolated after a patient with a splenic abscess was treated with ciprofloxacin[21]. One of the first studies done in vitro looked at the resistance profiles of Salmonella spp. after repeated exposure to FQs and β-lactams. They found that mutants generated after treatments with various FQs showed reduced susceptibility to a wide range of the antibiotics tested (seven different antibiotics). This was in contrast to mutants generated under treatment with β-lactams, which showed reduced susceptibility to fewer antibiotics (five antibiotics)[22]. Similarly, it has been shown that the treatment of methicillin-resistant St. aureus with sublethal FQ further enhances methicillin resistance[12]. A similar effect is seen with E. coli, as cells become resistant to quinolone and non-quinolone antibiotics after exposure to sublethal FQ[23]. Alongside these studies that show increases in resistance to non-quinolone antibiotics after FQ treatment, there is also evidence that sublethal treatment with quinolones increases mutation rates, mutation frequencies and recombination.[13][14][15][16][17][18]
2.2. Sublethal FQ Treatment Increases Mutation Rate
Quinolones are potent inducers of the SOS response[24][25][26][27]. As mentioned above, the SOS response is the bacterial response to DNA damage. It is regulated by RecA, a recombinase that is activated when there is DNA damage, and LexA, the repressor of the SOS regulon that autocleaves when RecA is active. The autocleavage of LexA results in the derepression of the SOS regulon that controls the expression of ~50 genes in E. coli, including three error-prone polymerases (PoI II, Pol IV and Pol V) that can introduce mutations[28]. Thus, it is not that surprising that quinolones could potentially increase the mutation rate. Indeed, many studies have shown that after the treatment with sublethal concentrations of FQs, there is an increase in mutation rate and mutation frequency.[12][13][14][15][16][17][27][29][30][31][32]
Ysern et al.[27] were the first to show that the quinolones increase mutagenesis through the induction of the SOS response. They suggested that the mutagenic effect was through the upregulation of Pol V. In 2005, Gillespie et al.[17] demonstrated that the treatment of Mycobacterium fortuitum with sub-MIC ciprofloxacin was able to increase the mutation rate by 72–120-fold. A more systematic study of the role of SOS in the mutagenic effect of quinolones was performed by Cirz et al.[29]. Using a neutropenic murine thigh infection model, they found that, in pathogenic E. coli, LexA, the repressor of the SOS response, was required for the evolution of the resistance induced by treatment with ciprofloxacin. They concluded that the homologous recombination pathway was important in the repair of ciprofloxacin-induced DNA damage and that LexA cleavage was induced during the repair. This then caused the upregulation of the three error-prone polymerases, which, together, generated the mutations that conferred resistance[29]. The same group went on to show that this was also the case in P. aeruginosa[16] and St. aureus.[15] LexA, RecA and error-prone DNA polymerases were also found to be upregulated in M. tuberculosis when treated with sublethal doses of ciprofloxacin[18]. Some groups, however, have suggested that upregulation of the error-prone polymerases is not the only mutagenic pathway induced by treatments with sublethal quinolones. Song et al.[33] showed that, when all three of the error-prone polymerases were deleted, ciprofloxacin-induced deletions still occurred. Long et al.[34] suggested that the increased mutagenesis observed with norfloxacin was not only due to the error-prone polymerases but, also, by indirect effects of the antibiotic on the mismatch-repair system and DNA-oxidative repair mechanisms. Oxidative stress has also been suggested as the main mutagenic pathway of a range of antibiotics, including norfloxacin, by Kohanski et al.[8]. They demonstrated that treating E. coli MG1655 with sublethal concentrations of norfloxacin in vitro caused multidrug resistance, which was abolished by the use of the ROS scavenger thiourea.
2.3. Sublethal FQ Treatment Stimulates Recombination
Sub-MIC treatment with quinolones have also been shown to increase genetic recombination. Ciprofloxacin has been demonstrated to stimulate homologous recombination in E. coli[13]. This was shown to be RecA-dependent and only partially reliant on induction of the SOS response; an uncleavable LexA mutant reduced the recombination but did not abolish it[13]. Sublethal ciprofloxacin was also demonstrated to induce nonhomologous recombination. This recombination was surprisingly independent of the SOS response but did require the other recombination pathways, RecBCD or RecFOR[14]. This study also looked at the effect of sublethal ciprofloxacin on conjugative transfer. Although they found no significant increase in the transfer of a conjugative plasmid, they did see an increase in horizontal gene transfer of an antibiotic-resistance gene from the plasmid to the genome[14]. Along these lines, three different FQs were shown to stimulate generalised transduction in Sa. Typhimurium. This was demonstrated through the transfer of a kanamycin resistance gene from a multdrug-resistant strain to susceptible strains through a P22-like bacteriophage[35].
2.4. Sublethal FQ Stimulates the Formation of Persisters
Another factor is how sublethal FQs has been shown to increase the presence of persister cells in the population[36][37][38][39][40]. Persisters are cells that can survive lethal concentrations of antibiotics due to phenotypic (but not genetic) changes. How persisters manage to survive antibiotic action is not clear, though it has been suggested that they do it by inactivating the drug target or lowering the drug uptake[41]. Persister cells have been demonstrated to form as a result of induction of the SOS response[36][38][39] or through a cellular response to starvation[40]. This persister population has also been shown to allow long-term survival to the exposure to the quinolone by allowing mutations to accumulate within the population that are then selected for by the antibiotic[36][37].
How sublethal FQs induce mutation appears to be complex, much like the lethality of quinolones and the repair of quinolone-induced DNA damage. There appear to be several pathways that lead to an increase in mutation. However, mutation seems to be a consequence of repair, and this mutagenesis does not increase the evolvability of the bacteria[42]. More work is needed to understand the role FQs play in mutagenesis, as a better understanding of how these drugs induce mutation may enable strategies to be put in place that will reduce their role in the acquisition of antibiotic resistance.
References
- Lesher, G.Y.; Froelich, E.J.; Gruett, M.D.; Bailey, J.H.; Brundage, R.P. 1,8-Naphthyridone derivatives. A new class of chemotherapeutic agents. J. Med. Pharm. Chem. 1962, 91, 1063–1065.
- Bisacchi, G.S. Origins of the quinolone class of antibacterials: An expanded “Discovery Story”. J. Med. Chem. 2015, 58, 4874–4882.
- Gillespie, S.H. The role of moxifloxacin in tuberculosis therapy. ERR 2016, 25, 19–28.
- Oliphant, C.M.; Green, G.M. Quinolones: A comprehensive review. Am. Fam. Physician 2002, 65, 455–464.
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574.
- EMEA/H/A-31/1452. Quinolone-and Fluoroquinolone-Containing Medicinal Products; European Commision: Amsterdam, The Netherlands, 2018.
- Jurado, S.; Medina, A.; de la Fuente, R.; Ruiz-Santa-Quiteria, J.A.; Orden, J.A. Resistance to non-quinolone antimicrobials in commensal Escherichia coli isolates from chickens treated orally with enrofloxacin. Jpn. J. Vet. Res. 2015, 63, 195–200.
- Kohanski, M.A.; DePristo, M.A.; Collins, J.J. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 2010, 37, 311–320.
- Pereira, R.V.; Siler, J.D.; Ng, J.C.; Davis, M.A.; Grohn, Y.T.; Warnick, L.D. Effect of on-farm use of antimicrobial drugs on resistance in fecal Escherichia coli of preweaned dairy calves. J. Dairy Sci. 2014, 97, 7644–7654.
- Chuppava, B.; Keller, B.; El-Wahab, A.A.; Meißner, J.; Kietzmann, M.; Visscher, C. Resistance of Escherichia coli in turkeys after therapeutic or environmental exposition with enrofloxacin depending on flooring. Int. J. Environ. Res. Public Health 2018, 15, 1993.
- Li, J.; Hao, H.; Dai, M.; Zhang, H.; Ning, J.; Cheng, G.; Shabbir, M.A.B.; Sajid, A.; Yuan, Z. Resistance and virulence mechanisms of Escherichia coli selected by Enrofloxacin in Chicken. Antimicrob. Agents Chemother. 2019, 63, e01824-18.
- Tattevin, P.; Basuino, L.; Chambers, H.F. Subinhibitory fluoroquinolone exposure selects for reduced beta-lactam susceptibility in methicillin-resistant Staphylococcus aureus and alterations in the SOS-mediated response. Res. Microbiol. 2009, 160, 187–192.
- Lopez, E.; Blazquez, J. Effect of subinhibitory concentrations of antibiotics on intrachromosomal homologous recombination in Escherichia coli. Antimicrob. Agents Chemother. 2009, 53, 3411–3415.
- Lopez, E.; Elez, M.; Matic, I.; Blazquez, J. Antibiotic-mediated recombination: Ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli. Mol. Microbiol. 2007, 64, 83–93.
- Cirz, R.T.; Jones, M.B.; Gingles, N.A.; Minogue, T.D.; Jarrahi, B.; Peterson, S.N.; Romesberg, F.E. Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J. Bacteriol. 2007, 189, 531–539.
- Cirz, R.T.; O’Neill, B.M.; Hammond, J.A.; Head, S.R.; Romesberg, F.E. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J. Bacteriol. 2006, 188, 7101–7110.
- Gillespie, S.H.; Basu, S.; Dickens, A.L.; O’Sullivan, D.M.; McHugh, T.D. Effect of subinhibitory concentrations of ciprofloxacin on Mycobacterium fortuitum mutation rates. J. Antimicrob. Chemother. 2005, 56, 344–348.
- O’Sullivan, D.M.; Hinds, J.; Butcher, P.D.; Gillespie, S.H.; McHugh, T.D. Mycobacterium tuberculosis DNA repair in response to subinhibitory concentrations of ciprofloxacin. J. Antimicrob. Chemother. 2008, 62, 1199–1202.
- Marshall, B.M.; Levy, S.B. Food animals and antimicrobials: Impacts on human health. Clin. Microbiol. Rev. 2011, 24, 718–733.
- Lee, S.J.; Park, N.H.; Mechesso, A.F.; Lee, K.J.; Park, S.C. The phenotypic and molecular resistance induced by a single-exposure to sub-mutant prevention concentration of marbofloxacin in Salmonella Typhimurium isolates from swine. Vet. Microbiol. 2017, 207, 29–35.
- Pers, C.; Søgaard, P.; Pallesen, L. Selection of multiple resistance in Salmonella enteritidis during treatment with ciprofloxacin. Scand. J. Infect. Dis. 1996, 28, 529–531.
- Cebrian, L.; RodrÍGuez, J.C.; Escribano, I.; Royo, G. Effect of exposure to fluoroquinolones and beta-lactams on the in vitro activity of other groups of antibiotics in Salmonella spp. APMIS 2006, 114, 523–528.
- Ching, C.; Zaman, M.H. Development and selection of low-level multi-drug resistance over an extended range of sub-inhibitory ciprofloxacin concentrations in Escherichia coli. Sci. Rep. 2020, 10, 8754.
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392.
- Piddock, L.J.V.; Wise, R. Induction of the SOS response in Escherichia coli by 4-quinolone antimicrobial agents. FEMS Microbiol. Lett. 1987, 41, 289–294.
- Lewin, C.S.; Howard, B.M.A.; Ratcliff, N.T.; Smith, J.T. 4-Quinolones and the SOS response. J. Med. Microbiol. 1989, 29, 139–144.
- Ysern, P.; Clerch, B.; Castańo, M.; Gibert, I.; Barbé, J.; Llagostera, M. Induction of SOS genes in Escherichia coli and mutagenesis in Salmonella typhimurium by fluoroquinolones. Mutagenesis 1990, 5, 63–66.
- Kreuzer, K.N. DNA damage responses in prokaryotes: Regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012674.
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crecy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005, 3, e176.
- Cirz, R.T.; Romesberg, F.E. Induction and inhibition of ciprofloxacin resistance-conferring mutations in hypermutator bacteria. Antimicrob. Agents Chemother. 2006, 50, 220–225.
- Thi, T.D.; López, E.; Rodríguez-Rojas, A.; Rodríguez-Beltrán, J.; Couce, A.; Guelfo, J.R.; Castañeda-García, A.; Blázquez, J. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J. Antimicrob. Chemother. 2011, 66, 531–538.
- Blazquez, J.; Rodriguez-Beltran, J.; Matic, I. Antibiotic-induced genetic variation: How it arises and how it can be prevented. Annu. Rev. Microbiol. 2018, 72, 209–230.
- Song, L.Y.; Goff, M.; Davidian, C.; Mao, Z.; London, M.; Lam, K.; Yung, M.; Miller, J.H. Mutational consequences of ciprofloxacin in Escherichia coli. Antimicrob. Agents Chemother. 2016, 60, 6165–6172.
- Long, H.; Miller, S.F.; Strauss, C.; Zhao, C.; Cheng, L.; Ye, Z.; Griffin, K.; Te, R.; Lee, H.; Chen, C.-C.; et al. Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc. Natl. Acad. Sci. USA 2016, 113, E2498–E2505.
- Bearson, B.L.; Brunelle, B.W. Fluoroquinolone induction of phage-mediated gene transfer in multidrug-resistant Salmonella. Int. J. Antimicrob. Agents 2015, 46, 201–204.
- Dörr, T.; Lewis, K.; Vulić, M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 2009, 5, e1000760.
- Barrett, T.C.; Mok, W.W.K.; Murawski, A.M.; Brynildsen, M.P. Enhanced antibiotic resistance development from fluoroquinolone persisters after a single exposure to antibiotic. Nat. Commun. 2019, 10, 1177.
- Goneau, L.W.; Yeoh, N.S.; MacDonald, K.W.; Cadieux, P.A.; Burton, J.P.; Razvi, H.; Reid, G. Selective target inactivation rather than global metabolic dormancy causes antibiotic tolerance in uropathogens. Antimicrob. Agents Chemother. 2014, 58, 2089–2097.
- Theodore, A.; Lewis, K.; Vulić, M. Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway. Genetics 2013, 195, 1265–1276.
- Gutierrez, A.; Jain, S.; Bhargava, P.; Hamblin, M.; Lobritz, M.A.; Collins, J.J. Understanding and sensitizing density-dependent persistence to quinolone antibiotics. Mol. Cell 2017, 68, 1147–1154.e3.
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464.
- Torres-Barceló, C.; Kojadinovic, M.; Moxon, R.; MacLean, R.C. The SOS response increases bacterial fitness, but not evolvability, under a sublethal dose of antibiotic. Proc. R. Soc. B 2015, 282, 20150885.



