 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | David Gonzalez-Serna | + 2763 word(s) | 2763 | 2020-12-15 07:35:33 | | | |
| 2 | Dean Liu | -1466 word(s) | 1297 | 2020-12-22 03:15:58 | | |
Video Upload Options
Immune-mediated diseases (IMDs) are complex pathologies that are strongly influenced by environmental and genetic factors. Associations between genetic loci and susceptibility to these diseases have been widely studied, and hundreds of risk variants have emerged during the last two decades, with researchers observing a shared genetic pattern among them.
1. Introduction
Immune mediated diseases (IMDs) are a diverse group of pathologies with different etiologies, characterized by a dysregulation of the immune system. These diseases show different effects on the organism, including either systemic or local symptoms, which may overlap between the diseases[1]. This complexity makes their diagnosis a clinical challenge, as different IMDs are found to have shared comorbidities and may co-occur in the same patient. A common example is cardiovascular disorders, which are present in several of these diseases[2][3][4][5], as well as the presence of autoantibodies, which have great clinical and diagnostic significance[6][7].
Thus, the high rates of familial clustering and comorbidities observed across IMDs indicate that they share molecular mechanisms of disease pathogenesis[8]. In the last decades, large-scale genetic studies, such as genome-wide association studies (GWAS) and Immunochip studies[9][10] have been essential to our understanding of IMD genetics, allowing the identification of a considerable number of loci associated with each individual disease [11][12][13][14], but also suggesting the existence of a common genetic background in autoimmunity[8].
However, despite the success of GWAS, most of the polymorphisms associated with IMDs are located in non-coding regulatory regions of the genome and therefore their direct consequences on the disease are not clear[15][16]. In this regard, functional genomics is very useful in order to identify the mechanism of action of disease-associated variants and therefore the mechanisms underlying complex diseases, thus allowing progress in translating genetic findings to the clinic[17][18]. As shown in Figure 1, the process from the identification of a disease-associated variant to its characterization at the phenotypic level includes different functional assessments, which differ depending on the genomic location of the variant.
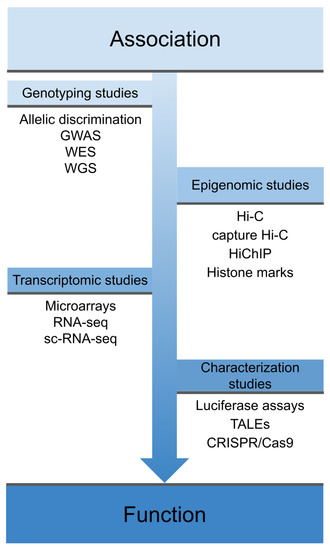
Figure 1. Overview of different techniques used in functional genomics. These techniques cover the spectrum from early association studies, through techniques that explore the physical interaction of these variants and their effects on the transcriptome, to phenotype characterization and gene function. GWAS: genome-wide association studies; WES: whole-exome sequencing; WGS: whole-genome sequencing; sc-RNA-seq: single-cell RNA sequencing, TALEs: Transcription activator-like effectors; CRISPR: clustered regularly interspaced short palindromic repeats; Cas9: CRISPR associated protein 9.
2. Shared Genetics across IMDs
In recent years, many efforts have been made in order to identify risk loci shared among IMDs by combining GWAS or Immunochip data across several diseases. This strategy allows a direct comparison of the genetic component of these diseases, as well as an increase in statistical power to detect associations with low-effect variants. To date, a considerable amount of pairwise cross-disease meta-analyses of GWAS data of systemic rheumatic diseases has been published[19][20][21], which has led to the identification of many risk loci shared between pairs of these diseases. Furthermore, five studies combining the GWAS or Immunochip data of multiple IMDs simultaneously have been published, thus identifying a total of 75 new shared risk loci with some degree of pleiotropy in autoimmunity, which could partially explain the comorbidity observed among IMDs[22][23][24][25][26]. Good examples of known shared risk loci in IMDs are PTPN22, IL23R and TNFAIP3, which have allowed the repositioning of anti-TNF and anti-IL-12/IL-23 therapies to be used in rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), among others[1].
On the other hand, a recent study identified shared germline variants that predispose patients to RA, SLE and primary Sjögren’s syndrome (SjS) through whole-exome sequencing of 31 families, highlighting related T-cell-activating genes[27]. This familial aggregation suggests that a specific molecular pattern, leading to common pathogenesis in certain IMDs, could exist. Furthermore, Li et al. [22] quantified pairwise genetic sharing across 17 IMDs from the Immunobase resource, revealing a closer association among major systemic IMDs (including RA, SLE and systemic sclerosis (SSc)) than with other autoimmune disorders, such as psoriasis and inflammatory bowel disease. These studies support the idea that genetic pathways are shared among apparently clinically different IMDs and therefore a molecular reclassification of these diseases could lead to the discovery of new biomarkers for patient stratification and prognosis[28]. In line with this, a recent study stratified seven systemic IMDs into groups of molecular patterns, taking into account high dimensional molecular data including genome, transcriptome, and methylome data from whole blood samples, performing an unsupervised clustering analysis. Authors observed that systemic IMDs clustered in three different groups, representing “inflammatory”, “lymphoid” and “interferon” groups, with specific molecular patterns independently from their clinical classification[29].
The emergence of GWAS and associated genotyping platforms led to an increase in the number of variants associated with complex diseases, allowing for the development of more accurate genomic risk score (GRS) calculations, a direct application of genomic data to the clinical setting. GRS measures the additive effect of single nucleotide polymorphisms (SNPs), calculating the relative risk of individuals suffering from a given disease [30][31][32]. GRSs have been applied to different IMDs such as SLE, RA, SSc and psoriatic arthritis (PsA)[33][34][35][36]. Furthermore, genomic data can be useful for making a differential diagnosis, which is especially interesting in the case of inflammatory arthritis, since these conditions present with similar symptoms in the early stages[37]. In addition, the different relevant symptoms of the disease can be used to increase the predictive power of GRS, such as the appearance of lupus nephritis in SLE[33], or the appearance of autoantibodies in SSc[35].
3. New Approaches for Drug Targeting
The identification of new targets is a critical step in the drug discovery process. In this sense, the recent massive accumulation of different genomic data, mainly through GWAS, and their annotation through different functional data, establish the perfect framework to elucidate the underlying pathogenic mechanisms of IMDs and thus prioritize potential new therapeutic targets[38].
In this regard, one of the main examples of the usefulness of genomic studies in the identification of potential new drug targets is a study published by Okada et al.[11]. Through the largest GWAS conducted in RA, the authors identified more than 100 loci associated with this disorder. Subsequently, using bioinformatic methods based on functional annotation, they identified a total of 98 biological candidate genes, some of which were targets of RA therapies, whereas others were targets of drugs potentially useful for the treatment of this disease. A more recent GWAS performed in SSc identified a total of 27 independent risk loci for this condition. The subsequent functional analysis, using HiChIP data, of the most probable causal variants allowed the identification of 43 robust target genes, highlighting CD80 and BLK as potential drug targets[14]. Furthermore, meta-analysis of GWAS data, including different IMDs, also led to the identification of shared risk loci, as well as potential new drug targets through drug enrichment analysis[24]. It is worth mentioning that drugs with mechanisms based on genetic evidence have a higher probability to be approved through the drug development process [39].
On the other hand, the biological function of a genetic change can be easy to observe, thus making it easy to identify a potential drug target. For example, the knowledge of mutations and deletions occurring in the JAK3 gene that cause severe combined immunodeficiency[40]was useful to develop drugs such as tofacitinib to treat inflammation in RA[41].
Additionally, genomic and epigenomic data, using functional genomic approaches, are being integrated in order to facilitate drug repurposing in IMDs[42]. In this regard, analysis of capture Hi-C data from B-cells and T-cells to identify causal genes for rheumatic diseases revealed many disease-associated genes to be targets of existing drugs. A recent study demonstrated an approach that integrates functional genomics and immune-mediated annotations with the evidence of physical interaction in order to prioritize drug targets, an approach which has been validated and applied in RA[43]. In this sense, the emergence of TNF as a highly ranked target confirms the utility of this approach [43][44].
References
- Cho, J.H.; Feldman, M. Heterogeneity of autoimmune diseases: Pathophysiologic insights from genetics and implications for new therapies. Nat. Med. 2015, 21, 730–738.
- López-Mejías, R.; Castañeda, S.; González-Juanatey, C.; Corrales, A.; Ferraz-Amaro, I.; Genre, F.; Remuzgo-Martínez, S.; Rodriguez-Rodriguez, L.; Blanco, R.; Llorca, J.; et al. Cardiovascular risk assessment in patients with rheumatoid arthritis: The relevance of clinical, genetic and serological markers. Autoimmun. Rev. 2016, 15, 1013–1030.
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888.
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699.
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038.
- Pisetsky, D.S.; Lipsky, P.E. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2020, 16, 565–579.
- Bournia, V.-K.; Vlachoyiannopoulos, P.G. Subgroups of Sjögren syndrome patients according to serological profiles. J. Autoimmun. 2012, 39, 15–26.
- Parkes, M.; Cortes, A.; van Heel, D.A.; Brown, M.A. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet. 2013, 14, 661–673.
- Polychronakos, C. Fine points in mapping autoimmunity. Nat. Genet. 2011, 43, 1173–1174.
- Cortes, A.; Brown, M.A. Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 2011, 13, 101.
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381.
- Bentham, J.; Morris, D.L.; Cunninghame Graham, D.S.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martín, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464.
- Carmona, F.D.; Vaglio, A.; Mackie, S.L.; Hernández-Rodríguez, J.; Monach, P.A.; Castañeda, S.; Solans, R.; Morado, I.C.; Narváez, J.; Ramentol-Sintas, M.; et al. A genome-wide association study identifies risk alleles in plasminogen and P4HA2 associated with giant cell arteritis. Am. J. Hum. Genet. 2017, 100, 64–74.
- López-Isac, E.; Acosta-Herrera, M.; Kerick, M.; Assassi, S.; Satpathy, A.T.; Granja, J.; Mumbach, M.R.; Beretta, L.; Simeón, C.P.; Carreira, P.; et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 2019, 10, 4955.
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22.
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730.
- Ding, J.; Frantzeskos, A.; Orozco, G. Functional genomics in autoimmune diseases. Hum. Mol. Genet. 2020, 29, R59–R65.
- Zeggini, E.; Gloyn, A.L.; Barton, A.C.; Wain, L.V. Translational genomics and precision medicine: Moving from the lab to the clinic. Science 2019, 365, 1409–1413.
- Martin, J.-E.; Assassi, S.; Diaz-Gallo, L.-M.; Broen, J.C.; Simeon, C.P.; Castellvi, I.; Vicente-Rabaneda, E.; Fonollosa, V.; Ortego-Centeno, N.; González-Gay, M.A.; et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum. Mol. Genet. 2013, 22, 4021–4029.
- López-Isac, E.; Martín, J.-E.; Assassi, S.; Simeón, C.P.; Carreira, P.; Ortego-Centeno, N.; Freire, M.; Beltrán, E.; Narváez, J.; Alegre-Sancho, J.J.; et al. Brief report: IRF4 newly identified as a common susceptibility locus for systemic sclerosis and rheumatoid arthritis in a cross-disease meta-analysis of genome-wide association studies. Arthritis Rheumatol. 2016, 68, 2338–2344.
- Márquez, A.; Vidal-Bralo, L.; Rodríguez-Rodríguez, L.; González-Gay, M.A.; Balsa, A.; González-Álvaro, I.; Carreira, P.; Ortego-Centeno, N.; Ayala-Gutiérrez, M.M.; García-Hernández, F.J.; et al. A combined large-scale meta-analysis identifies COG6 as a novel shared risk locus for rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 286–294.
- Li, Y.R.; Li, J.; Zhao, S.D.; Bradfield, J.P.; Mentch, F.D.; Maggadottir, S.M.; Hou, C.; Abrams, D.J.; Chang, D.; Gao, F.; et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med. 2015, 21, 1018–1027.
- Ellinghaus, D.; Jostins, L.; Spain, S.L.; Cortes, A.; Bethune, J.; Han, B.; Park, Y.R.; Raychaudhuri, S.; Pouget, J.G.; Hübenthal, M.; et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 2016, 48, 510–518.
- Márquez, A.; Kerick, M.; Zhernakova, A.; Gutierrez-Achury, J.; Chen, W.-M.; Onengut-Gumuscu, S.; González-Álvaro, I.; Rodriguez-Rodriguez, L.; Rios-Fernández, R.; González-Gay, M.A.; et al. Meta-analysis of immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med. 2018, 10, 97.
- Ortiz-Fernández, L.; Carmona, F.D.; López-Mejías, R.; González-Escribano, M.F.; Lyons, P.A.; Morgan, A.W.; Sawalha, A.H.; Merkel, P.A.; Smith, K.G.C.; González-Gay, M.A.; et al. Cross-phenotype analysis of immunochip data identifies KDM4C as a relevant locus for the development of systemic vasculitis. Ann. Rheum. Dis. 2018, 77, 589–595.
- Acosta-Herrera, M.; Kerick, M.; González-Serna, D.; Wijmenga, C.; Franke, A.; Gregersen, P.K.; Padyukov, L.; Worthington, J.; Vyse, T.J.; Alarcón-Riquelme, M.E.; et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann. Rheum. Dis. 2019, 78, 311–319.
- Wang, Y.; Chen, S.; Chen, J.; Xie, X.; Gao, S.; Zhang, C.; Zhou, S.; Wang, J.; Mai, R.; Lin, Q.; et al. Germline genetic patterns underlying familial rheumatoid arthritis, systemic lupus erythematosus and primary Sjögren’s syndrome highlight T cell-initiated autoimmunity. Ann. Rheum. Dis. 2020, 79, 268–275.
- Barturen, G.; Beretta, L.; Cervera, R.; Van Vollenhoven, R.; Alarcón-Riquelme, M.E. Moving towards a molecular taxonomy of autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 180.
- Barturen, G.; Babaei, S.; Català-Moll, F.; Martínez-Bueno, M.; Makowska, Z.; Martorell-Marugán, J.; Carmona-Sáez, P.; Toro-Domínguez, D.; Carnero-Montero, E.; Teruel, M.; et al. Integrative Analysis Reveals a Molecular Stratification of Systemic Autoimmune Diseases. Arthritis Rheumatol. 2020.
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224.
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590.
- Choi, S.W.; Mak, T.S.-H.; O’Reilly, P.F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 15, 2759–2772.
- Chen, L.; Wang, Y.-F.; Liu, L.; Bielowka, A.; Ahmed, R.; Zhang, H.; Tombleson, P.; Roberts, A.L.; Odhams, C.A.; Cunninghame Graham, D.S.; et al. Genome-wide assessment of genetic risk for systemic lupus erythematosus and disease severity. Hum. Mol. Genet. 2020, 29, 1745–1756.
- Stahl, E.A.; Wegmann, D.; Trynka, G.; Gutierrez-Achury, J.; Do, R.; Voight, B.F.; Kraft, P.; Chen, R.; Kallberg, H.J.; Kurreeman, F.A.S.; et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat. Genet. 2012, 44, 483–489.
- Bossini-Castillo, L.; Villanueva-Martin, G.; Kerick, M.; Acosta-Herrera, M.; López-Isac, E.; Simeón, C.P.; Ortego-Centeno, N.; Assassi, S.; International SSc Group; Australian Scleroderma Interest Group (ASIG); et al. Genomic risk score impact on susceptibility to systemic sclerosis. Ann. Rheum. Dis. 2020.
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166.
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860.
- Russell, S.M.; Tayebi, N.; Nakajima, H.; Riedy, M.C.; Roberts, J.L.; Aman, M.J.; Migone, T.-S.; Noguchi, M.; Markert, M.L.; Buckley, R.H.; et al. Mutation of Jak3 in a patient with SCID: Essential role of jak3 in lymphoid development. Science 1995, 270, 797–800.
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860.
- Russell, S.M.; Tayebi, N.; Nakajima, H.; Riedy, M.C.; Roberts, J.L.; Aman, M.J.; Migone, T.-S.; Noguchi, M.; Markert, M.L.; Buckley, R.H.; et al. Mutation of Jak3 in a patient with SCID: Essential role of jak3 in lymphoid development. Science 1995, 270, 797–800.
- Van Vollenhoven, R.F.; Fleischmann, R.; Cohen, S.; Lee, E.B.; García Meijide, J.A.; Wagner, S.; Forejtova, S.; Zwillich, S.H.; Gruben, D.; Koncz, T.; et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 508–519.
- Kingsmore, K.M.; Grammer, A.C.; Lipsky, P.E. Drug repurposing to improve treatment of rheumatic autoimmune inflammatory diseases. Nat. Rev. Rheumatol. 2020, 16, 32–52.
- Fang, H.; ULTRA-DD Consortium; De Wolf, H.; Knezevic, B.; Burnham, K.L.; Osgood, J.; Sanniti, A.; LledóLara, A.; Kasela, S.; De Cesco, S.; et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 2019, 51, 1082–1091.
- Fang, H.; Chen, L.; Knight, J.C. From genome-wide association studies to rational drug target prioritisation in inflammatory arthritis. Lancet Rheumatol. 2020, 2, e50–e62.


