 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Javier Oroz | + 3405 word(s) | 3405 | 2020-12-17 05:10:06 | | | |
| 2 | Camila Xu | Meta information modification | 3405 | 2020-12-23 02:32:54 | | |
Video Upload Options
Hsp70, Hsp90, and their co-chaperones are crucial members of the proteostasis network that are able to recognize misfolded proteins, aberrant condensates and protein aggregates, triaging proteins for refolding or degradation. These members of the chaperome are considered major sentinels impeding the molecular processes that lead to cell damage in the course of degenerative proteinopathies. Indeed, Hsp70, Hsp90 and their co-chaperones are increasingly recognized as therapeutic targets for the development of treatments against prevalent protein misfolding diseases.
1. Dynamic Hsp70 and Hsp90 Are Closely Monitored by Co-Chaperones
The Hsp70 family of molecular chaperones is ubiquitously expressed and particularly relevant in the early stages of nascent polypeptide folding [1][2]. Human Hsp70 presents a variety of highly identical isoforms that coexist in cellular compartments, yet show functional specificity [3]. Hsp70 is composed of two domains, a 40 kDa, actin-like N-terminal nucleotide-binding domain (NBD) that binds ATP and regulates allostery and a 25 kDa C-terminal substrate-binding domain (SBD) (Figure 1). The substrate-binding pocket present in the SBD is capped by a lid, which undergoes large conformational changes following the ATP hydrolysis cycle toward the opening of the binding pocket and the release of the substrate (Figure 1).
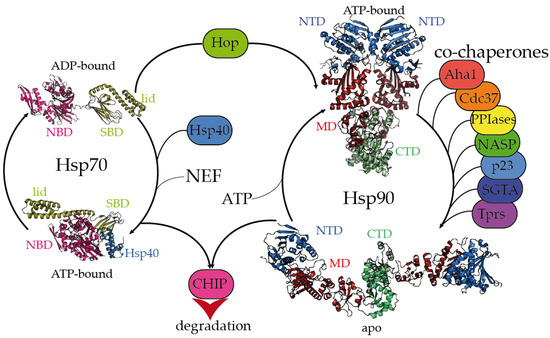
Figure 1. Hsp70 and Hsp90 chaperones are tightly regulated by co-chaperones. Hsp70 undergoes large allosteric changes upon ATP hydrolysis and substrate binding, from an extended, ADP-bound conformation with the substrate-binding domain (SBD) closed by the lid (PDB code 2kho) [4] to a collapsed, ATP-bound conformation with the SBD opened, favoring substrate release (PDB code 5nro) [5]. NBD stands for the nucleotide-binding domain. Hsp40 regulates the substrate folding and ATP hydrolysis by Hsp70 (Hsp40 J domain is included in PDB 5nro in the blue ribbon representation). NEF stands for the nucleotide exchange factor. Hop (Hsp-organizing protein) promotes the transfer of the substrate from the Hsp70 machinery to Hsp90. Hsp90 coexists in several conformations, from an extended, apo conformation [6] to a closed, nucleotide bound conformation, where the nucleotide-binding domains (NTD) would rotate to promote ATP hydrolysis [7]. CTD and MD stand for the C-terminal and middle domains, respectively. Hsp90′s activation cycle is closely regulated by co-chaperones [7]. Several representative human co-chaperones are included in the figure and are reviewed elsewhere [8][9]. Tprs stands for TPR-containing proteins. Phosphorylation of Hsp70 and Hsp90 C-terminal tails dictate the binding of CHIP, promoting substrate degradation [10].
While the inherent ATPase activity of human Hsp70 is significantly slow, it is stimulated by the Hsp40/DNAJ family upon interaction with the J-domain, which additionally aids Hsp70 in the selective recruitment of clients [11][12][3]. Hsp40/DNAJ proteins interact differently with Hsp70 chaperones [13]. In the major class B, DNAJB1 exhibits a class-dependent autoinhibitory mechanism, in which a Gly/Phe-rich segment blocks the Hsp70-binding sites located in the J-domain. The presence of an additional Hsp70-binding site, which is not present in the class A Hsp40s, releases the Gly-Phe inhibition upon binding to the C-terminal IEEVD tail of Hsp70 [13]. In Hsp70, the NBD and SBD domains are separated by a highly conserved, dynamic, and amphipathic linker, which is crucial for the allosteric communication between both domains [11][14][15]. The sequence conservation between isoforms dramatically decays in the C-terminal region of the lid subdomain that closes the SBD, and in the following short C-terminal disordered fragment [14]. Interestingly, these variable regions determine the differential effects of Hsp70 isoforms on the aggregation or degradation of the Alzheimer’s-disease-related protein tau [16]. The very last C-terminal sequence of Hsp70 (containing the sequence IEEVD) is specifically recognized by the tetratricopeptide repeats (TPRs) present in the co-chaperone Hop, which escorts Hsp70:client complexes toward Hsp90 [17].
The biologically active Hsp90 dimer exhibits a tightly regulated equilibrium of extended and closed conformations (Figure 1) [7][6]. The ATPase activity is located in the N-terminal domain (NTD), which is highly conserved among the different Hsp90 isoforms [18]. The NTD is followed by a highly flexible, negatively charged linker, which is particularly relevant for client recognition [6][19][20]. The middle domain (MD) is important for allostery and co-chaperone and client binding, and the C-terminal domain (CTD) is critical for the dimerization and stabilization of Hsp90:client complexes [6]. Similar to Hsp70, Hsp90 has a conserved MEEVD motif at its C-terminus, which is targeted by TPR-containing proteins [6][17]. Human Hsp90 participates in the proper folding, maturation, and maintenance of a vast number of clients [21][22], and its activity, selectivity, and functionality are regulated by co-chaperones. Interestingly, the close inspection of all the Hsp90:co-chaperone complex structures described so far has revealed that co-chaperones that intervene at different stages of the Hsp90 activation cycle [7] bind to different interfaces of Hsp90, which delimits the available sites for the interaction with clients [6][19][23][24][25][26][27].
2. Selective Recognition of Misfolded Proteins by Hsp70 and Hsp90
Because Hsp70 recognizes short, hydrophobic sequences, and the availability and exposure of such regions trigger protein aggregation [2][28], the misregulation of Hsp70 and its co-chaperones can be related to the progress of protein misfolding diseases [29][30][31][32]. Indeed, pharmacological upregulation or overexpression of Hsp70 preserves viability in cells expressing aggregation-prone proteins, either by blocking the formation of toxic species [33][34][35][36] or by inducing aggregation into innocuous deposits [37]. Such sequences targeted by Hsp70 can be exposed as the result of partial misfolding of stably folded protein domains into metastable intermediates or present in protein sequences that remain intrinsically disordered. For instance, the protein transthyretin (TTR) forms a functional tetramer but can aggregate into insoluble deposits in TTR amyloidosis [38], causing neuropathy and cardiomyopathy [39]. During tetramer dissociation and misfolding, the C-terminal β-strand in the monomer of TTR unfolds, revealing hydrophobic segments [40] in a process that can be reverted by the protective T119M mutation [41]. However, it is still unknown whether Hsp70 directly binds misfolded TTR or whether the interaction would be mediated via an Hsp40 co-chaperone [3]. Although TTR deposits are mainly extracellular, they induce the activation of intracellular Hsp70, possibly through stimulation of the inflammatory response [42]. In a recent study, Masser and co-workers elegantly showed how Hsp70 specifically binds and inhibits Hsf1 (the transcription factor that induces the heat-shock response upon folding stress to balance proteostasis) [43]. Interestingly, increasing amounts of misfolded peptides in the cytosol during stress would titrate out Hsp70, releasing and activating Hsf1, which could ultimately lead to hyperstressed conditions and proteostasis failure [43].
The recognition of misfolded protein conformations by Hsp70 is not always thorough and may depend on the microenvironment. Hsp70 was found to bind specific conformations of human PrP prion protein in membrane microdomains in Drosophila, preventing the accumulation of misfolded conformers and alleviating PrP neurotoxicity [44]. Mutations in the superoxide dismutase 1 (SOD1) gene are related to familial cases of amyotrophic lateral sclerosis (ALS). Several mutations were found to reduce ion binding and promote partial misfolding in the cytosol [45]. One of these mutations, A4V, promotes the exposure of aggregation-prone regions, but not of Hsp70 recognition motifs (containing the sequences 64HFNPLSR70 and 114IGRTLVV120), providing an explanation for the accumulation of toxic SOD1 aggregates that result from incomplete recognition by Hsp70 [46][47]. The tumor suppressor p53 is another pertinent example of a misfolded target of Hsp70. Almost half of human cancers are associated with the loss of function and/or gain-of-toxic function of p53, which is triggered by its amyloid-like aggregation [48]. The Hsp70 recognition motif (251ILTII255 in the sequence of p53) is buried in the core of p53′s tridimensional structure [49][50]. The role of Hsp70 in regulating p53′s activity has remained controversial since some evidence indicates that Hsp70 induces p53 misfolding and cytoplasmic aggregation [51][52]. Interestingly, Hsp70 was found to trap misfolded intermediates of p53 with the assistance of Hsp40 co-chaperones, hence stabilizing aggregation-prone conformations [50]. The recruitment of Hsp90 and its co-chaperone Hop impedes the Hsp70-driven aggregation of p53 since Hop would promote the transfer of misfolded p53 to Hsp90. Hsp90 and Hop then restore the native state of p53 [50][53]. Therefore, proper recognition of misfolded motifs in pathogenic proteins by Hsp70 and its intricate coalition with Hsp90 and co-chaperones are key to preventing further aggregation and maintaining a balanced proteostasis.
Hsp70 recognition motifs remain more exposed in intrinsically disordered proteins (IDPs). The natively disordered protein tau plays multiple roles in neurons but can form aggregates of different compositions in tauopathies [54]. Despite being highly soluble, tau contains several hydrophobic fragments (containing the sequences 275VQIINK280 and 306VQIVYK311), which mediate its amyloid aggregation [54] and are specifically recognized by Hsp70 [16]. Intriguingly, only the inducible variant of Hsp70 is able to promote proteasomal tau clearance, which is mediated by the co-chaperone ubiquitin ligase CHIP [16]. Polyglutamine expansions in the protein huntingtin (HTT) form cytoplasmic inclusion bodies (huntingtin bodies) and are causative for hereditary forms of Huntington’s disease [55]. Hsp70 specifically recognizes the sequence ahead of the polyglutamine tract in HTT (containing the sequence 1MATLEKLMKAFESLKSF17), which mediates intermolecular associations that are relevant for aggregation [56] and may be important for stabilizing helical conformations within the polyglutamine tract [57]. Mediated by this interaction, Hsp70 and Hsp40 inhibit HTT aggregation and alleviate the derived toxicity [56]. Human islet amyloid polypeptide (IAPP) is the major component of the amyloid deposits found in patients with non-insulin-dependent (type II) diabetes mellitus. Bongiovanni and coworkers showed that a modified version of Hsp70 specifically targeted the sequence 56FGAILSS62 in IAPP [58]. Remarkably, Hsp70 seemed to first accelerate IAPP aggregation before suppressing it, leading to reduced cytotoxicity [58]. The TAR DNA binding protein of 43 kDa (TDP-43) is abundantly found forming insoluble aggregates in familial and sporadic cases of ALS, frontotemporal lobar degeneration, and even Alzheimer’s disease and the recently defined Limbic-predominant age-related TDP-43 encephalopathy (LATE) [59][60]. Overexpression of Hsp70 reduced TDP-43 aggregation without promoting its degradation [61]. Hsp70 and Hsp40 co-chaperones were found to stably interact with the C-terminal disordered region of TDP-43, preserving TDP-43′s solubility and functionality [62]. Similar to the Hsf1 inhibition mechanism mediated by Hsp70 mentioned earlier [43], activation of stress would enhance protein misfolding in the cytosol, which would titrate out Hsp70 and Hsp40 from TDP-43, favoring its aggregation and loss of function [62]. Overall, Hsp70 in combination with DNAJ/Hsp40 co-chaperones are very potent neutralizers of protein aggregation, particularly for IDPs, through a wide variety of mechanisms.
Evidence indicates that the interaction between Hsp90 and folded substrates are not restricted to a particular region of Hsp90 but cover a wide interface in the chaperone, leading to the formation of much more dynamic, pleomorphic, and multivalent complexes than in the case of Hsp70. For instance, in the absence of co-chaperones, Hsp90 forms multiple dynamic complexes with misfolded TTR and p53 [40][63][64]. Despite displaying selective recognition for misfolded TTR, Hsp90 was unable to promote the refolding of TTR in the absence of co-chaperones [40]. Conversely, the binding of Hsp90 promoted the interconversion between metastable conformers within an ensemble of conformations in p53 [63]. These dynamic modes of interactions are in apparent conflict with the structure of Hsp90 in a complex with the co-chaperone Cdc37 and the client Cdk4, which was solved using cryo-electron microscopy (cryo-EM) [19]. In that structure, Hsp90 traps a misfolded form of the client Cdk4 in a complex stabilized by the co-chaperone Cdc37. Therefore, questions are raised whether the presence of co-chaperones bound to Hsp90 would promote preferential modes of interaction for the client proteins. Nevertheless, evidence obtained from nuclear magnetic resonance (NMR) spectroscopy indicates that, in solution, co-chaperones bind to human Hsp90 in a very dynamic fashion, where fully bound forms of the Hsp90:co-chaperone complex coexist with a partially unbound co-chaperone [6][26].
The interaction between Hsp90 and IDP clients is highly dynamic and polymorphic. In the case of the longest isoform of human tau (called htau40 [65]), a large fraction of the protein binds to Hsp90, irrespective of the allosteric state of the chaperone [6]. Specifically, the proline-rich region and microtubule-binding domains of tau are strongly involved in the interaction with Hsp90. Interestingly, these regions are rich in positively charged residues, which may well be trapped by the negatively charged linker between the NTD and MD domains of Hsp90 [20]. In addition, the aggregation-prone, hydrophobic stretches present in tau are specifically recognized by nonpolar patches exposed along the surface of the different domains of Hsp90, supporting the multivalent nature of the interaction [6]. These hydrophobic stretches in tau, which were also specifically recognized by Hsp70 [16], adopt a partial β-structure in solution [65], which might be relevant for the recognition by chaperones (Figure 2). Hence, multiple conformations of tau adhere along one arm of the Hsp90 dimer in a strikingly polymorphic, yet specific manner [6]. Quite compellingly, the presence of a co-chaperone bound to Hsp90 drastically reshapes the tau conformational ensemble in the complex [6], thus leading to the presumption that studying the structural consequences of chaperone:co-chaperone complexes on bound clients may be more biologically relevant.
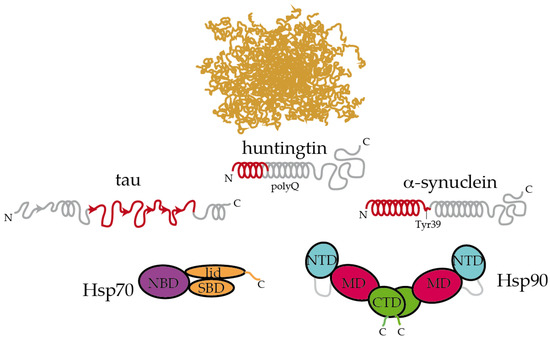
Figure 2. Hsp70 and Hsp90 recognize the structured moieties present in intrinsically disordered proteins (IDPs). A representative ensemble of conformations adopted by an IDP in solution is pictured at the top [66]. Tau, huntingtin, and α-synuclein contain elements that have a tendency to adopt a partial secondary structure (represented with helices and arrows for α-helices and β-strands, respectively) [65][67][68]. The moieties recognized by Hsp70 and Hsp90 are colored in red [6][16][69][68]. On tau, Hsp70 specifically recognizes the stretches 275VQIINK280, 306VQIVYK311, and 375KLTFRE380 [16]. The disordered C-terminal tails of Hsp70 and Hsp90 are represented. In Hsp90, the flexible, negatively charged linker connecting the NTD and the MD is represented with a grey line. The length of huntingtin is shortened for simplicity.
Inspecting the interaction between chaperones and IDPs reveals interesting insights. Extracellular senile plaques composed of amyloid-β (Aβ) peptide are also characteristic of Alzheimer’s disease [5]. Upon cleavage of the transmembrane amyloid precursor protein, amphipathic fragments of 40 and 42 residues (called Aβ40 and Aβ42) are released. Both Aβ40 and Aβ42 show structural transitions toward enrichment in a β-sheet structure and the propensity to aggregate [70]. Hsp90 and Hsp70:Hsp40 complexes could both interact with Aβ42 in the monomeric and oligomeric forms, triggering structural changes in Aβ42 that halt its further aggregation [71]. In a recent systematic study, Burmann and coworkers showed that several divergent chaperones, including Hsp70 and Hsp90, all recognized the same regions of monomeric α-synuclein [69]. α-Synuclein is the main constituent of the Lewy bodies, which are intracellular inclusions found in Parkinson’s disease [72]. More specifically, Hsp70 and Hsp90 recognized the N-terminal region containing the sequence 1MDVFMKGLSKAKEGVVAAAEKTKQGVAEAAGKTKE35, which has a tendency to adopt α-helical conformations [67], and the Tyr39 residue of α-synuclein. Interaction with Hsp70 and Hsp90 prevented α-synuclein’s oligomerization [69]. Interestingly, these interactions were found to be very transient in cells, and subtle changes in the cellular levels of α-synuclein or chaperones, modifications in α-synuclein or induction of cellular stress could dissociate the complexes and imbalance proteostasis, leading to α-synuclein aggregation and eventually causing Parkinson’s disease [69], In a similar fashion, Hsp90 specifically binds to the identical region of HTT that is recognized by Hsp70 [56][68]. This particular region, which was proposed to form an amphiphatic α-helix in solution is able to bind to multiple regions of Hsp90 [68].
Altogether, it appears that Hsp70 and Hsp90 recognize the same repertoire of hydrophobic stretches present in client proteins, with Hsp90 showing an additional affinity for bulky aromatic residues. Remarkably, when these chaperone recognition motifs are present in IDPs, they show a significant tendency to adopt a secondary structure (Figure 2). This raises the question of whether chaperone recognition is based on sequential or structural motifs. It is possible that Hsp70 and Hsp90 selectively bind to one side of the α-helix or β-strand where most of the hydrophobic sidechains of the motif reside [68]. Besides being rich in hydrophobic residues, the regions that are recognized by Hsp70 and Hsp90 on IDPs are also significantly abundant in positively charged residues [6]. Therefore, the interaction between chaperones and IDPs is multivalent [6]. The outcome of these apparently similar recognition mechanisms by Hsp70 and Hsp90 on the triaging of the client protein can be drastically different, but this resolution seems to be dictated by the specific co-chaperone recruited to the complex (Figure 1). For instance, the co-chaperones Hop and CHIP bind to the same regions of Hsp70 and Hsp90, and the selection is determined by phosphorylation of the binding site in the chaperones [10]. While the binding of Hop enhances client protein folding, the binding of CHIP promotes client degradation [10]. Thus, because Hsp90 is shown to be ambivalent in client triaging, it has been considered to play a passive scaffolder role in the context of protein misfolding diseases. Indeed, several co-chaperones have been raised as promising therapeutic targets for various neurodegenerative diseases [6][73][74][75][76].
3. Hsp70 and Hsp90 Play Crucial Roles in Disease-Related LLPS
LLPS is a long-studied physicochemical process whose enormous impact in protein biochemistry and cell biology has been recognized recently [77][78]. During the last decades, major cellular membraneless compartments covering a wide range of functions have been discovered [78][79][80][81]. These highly dynamic species are formed by the biomolecular coacervation of usually proteins and RNA to typically keep them dormant during stressful conditions [77], and the molecules within the condensate can be exchanged from the condensed to the dispersed phases. Proteinaceous LLPS is governed by weak, transient, and reversible interactions [82], and a pattern of “stickers” (aromatic residues) and “spacers” (polar moieties) on proteins has been established to determine the phase behavior [83][84]. Because of their elevated dynamics and plasticity and patterning of stickers and spacers, IDPs are actively involved in LLPS [84][84]. Intriguingly, even though biomolecular condensates are involved in a plethora of physiological functions, there is a delicate equilibrium between the physiological and aberrant condensates in diseases [85], and condensates that are formed by several proteins were shown to evolve to fibrillar solid aggregates of an amyloid nature [86][87][88][89][90].
Biomolecular condensates are metastable states that quickly react to changes in the environment. Thus, tight regulation is required to modulate condensates’ disassembly or their progress to fibrillar structures (either functional or pathogenic) [85][91][92]. In aging, when the stressful conditions lead to an overwhelmed PN [2], IDPs within condensates, such as RNP (ribonucleoprotein particle) granules or stress granules (SGs), engage misfolded proteins that result from defective ribosomal products [93]. In contrast, the promiscuous recruitment of misfolded proteins in the granules is avoided by the PN in normal conditions [93][94]. Surprisingly, while physiological SGs are devoid of misfolded proteins, and therefore, not recognized by the PN or autophagy machineries, aberrant SGs are rich in misfolded proteins, attracting members of the PN and autophagy machineries [95][96][94]. Along these lines, age-dependent decay in the activity of the PN [2] promotes the accumulation of aberrant SGs, as has indeed been observed in organisms of increasing age [97].
Rather than being cleared by autophagy, it appears that condensates are preferably disassembled by the PN, favoring the cellular recycling of the components [95]. Condensates, such as SGs, accumulate many different elements of the PN [95]. For instance, Hsp70 was found to be essential for dissolving SGs to reactivate translation in Drosophila after a heat shock [92]. In contrast, Hsp90 was found to be a key player for the formation and maintenance of P-bodies, which are cytoplasmic granules where inactive mRNAs and translation repressors are stored in a dormant state [98]. The inhibition of Hsp90 induces the disassembly of P-bodies, reactivating translation [98]. In addition, the inhibition of Hsp90 promotes the disassembly of TDP-43-containing SGs and pathogenic aggregates [99]. This apparent contradictory effect of Hsp70 versus Hsp90 in condensate maintenance and disassembly must be interpreted with caution since the pharmacological inhibition of Hsp90 usually induces the concomitant overactivation of Hsp70 and other sHsps [99][100][101]. The chaperone complex formed by Hsp70, the co-chaperone BAG3, and the sHsp Hspb8 is crucial for dissolving SGs and prevent the accumulation of aberrant granules in ALS [96][102]. In a sort of stepwise mechanism, Hsp70 promotes the rapid disassembly of SGs, where persisting aberrant SGs and remaining aggregates are transported through microtubules to protein inclusions for autophagic degradation [94].
It remains unclear how and which species are recognized by the Hsp70/Hsp90 machineries within the condensates. The lingering of misfolded proteins in high concentrations that are segregated either in the core or the periphery of the condensate [94][103] could promote structural conversions toward the acquisition of β-structure and cross-seeding processes as a starting point for amyloid formation [85], where these motifs could become targets for chaperone recognition. Audas and coworkers recently described the A-body, a nuclear condensate that arrests amyloidogenic proteins to languish in their aggregation in response to stressors [104]. Upon termination of the stress conditions, Hsp70 and Hsp90 chaperones disassemble the A-bodies and avoid the further aggregation of the constituents inside these condensates [104]. In addition, because aberrant SGs also enclose misfolded ribosomal products, improper regulation of SGs by the PN would not only promote the aggregation of amyloid proteins but also impair the synthesis of many proteins that are fundamental for the adaptation to aging due to the lack of function of these arrested ribosomal products [95]. Therefore, the misregulation of physiological condensates by the PN in aging can entail dramatic consequences, and deciphering the exact mechanisms of chaperone-mediated condensate dissolution or maintenance is imperative in this emerging field of research.
References
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, T.; Tugarinov, V.; Clore, G.M. Unraveling the structure and dynamics of the human DNAJB6b chaperone by NMR reveals insights into Hsp40-mediated proteostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 21529–21538. [Google Scholar] [CrossRef]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular mechanism of J-domain-triggered ATP hydrolysis by Hsp70 chaperones. Mol. Cell 2018, 69, 227–237. [Google Scholar] [CrossRef]
- Oroz, J.; Chang, B.J.; Wysoczanski, P.; Lee, C.-T.; Pérez-Lara, A.; Chakraborty, P.; Hofele, R.V.; Baker, J.D.; Blair, L.J.; Biernat, J.; et al. Structure and pro-toxic mechanism of the human Hsp90/PPIase/Tau complex. Nat. Commun. 2018, 9, 4532. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Molec. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.E.; Johnson, J.L. Human Hsp90 cochaperones: Perspectives on tissue-specific expression and identification of cochaperones with similar in vivo functions. Cell Stress Chaper. 2020, 1–11. Available online: https://link.springer.com/article/10.1007/s12192-020-01167-0 (accessed on 30 November 2020). [CrossRef]
- Peterson, L.B.; Blagg, B.S.J. To fold or not to fold: Modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283. [Google Scholar] [CrossRef]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.J.; Hrstka, R.; Lane, D.P.; Vojtesek, B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternative binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 2013, 32, 3101–3110. [Google Scholar] [CrossRef]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Faust, O.; Abayev-Avraham, M.; Wentik, A.S.; Maurer, M.; Nillegoda, N.B.; London, N.; Bukau, B.; Rosenzweig, R. HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature 2020, 587, 489–494. [Google Scholar] [CrossRef]
- Kabani, M.; Martineau, C.N. Multiple Hsp70 isoforms in the eukayoric cytosol: Mere redundancy or functional specificity? Curr. Genomics 2008, 9, 338–348. [Google Scholar] [CrossRef] [PubMed]
- English, C.A.; Sherman, W.; Meng, W.; Gierasch, L.M. The Hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J. Biol. Chem. 2017, 292, 14765–14774. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary, J.C., 3rd; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M.; et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Lott, A.; Oroz, J.; Zweckstetter, M. Molecular basis of the interaction of Hsp90 with its co-chaperone Hop. Protein Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef]
- Daturpalli, S.; Kniess, R.A.; Lee, C.T.; Mayer, M.P. Large rotation of the N-terminal domain of Hsp90 is important for interaction with some but not all client proteins. J. Mol. Biol. 2017, 429, 1406–1423. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Molec. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef]
- Meyer, P.; Prodromou, C.; Liao, C.; Hu, B.; Roe, S.M.; Vaughan, C.K.; Vlasic, I.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 2004, 23, 1402–1410. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucelotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kadota, Y.; Prodromou, C.; Shirasu, K.; Pearl, L.H. Structural basis for assembly of Hsp90-Sgt1-CHORD protein complexes: Implications for chaperoning of NLR innate immunity receptors. Mol. Cell 2010, 39, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Blair, L.J.; Zweckstetter, M. Dynamic Aha1 co-chaperone binding to human Hsp90. Prot. Sci. 2019, 28, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, M.; Myasnikov, A.G.; Elnatan, D.; Delaeter, N.; Nguyenquang, M.; Agard, D.A. Cryo-EM structures reveal a multistep mechanism of Hsp90 activation by co-chaperone Aha1. Biorxiv 2020. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Guo, H.; Yang, X.; Zhou, L.; Zhang, X.; Cheng, L.; Zeng, H.; Hu, F.B.; Tanguay, R.M.; Wu, T. Genetic variations in HSPA8 gene associated with coronary heart disease risk in a Chinese population. PLoS ONE 2010, 5, e9684. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.B.; Sommerville, R.B.; Allred, P.; Bell, S.; Ma, D.; Cooper, P.; Lopate, G.; Pestronk, A.; Weihl, C.C.; Baloh, R.H. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann. Neurol. 2012, 71, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Blumen, S.C.; Astord, S.; Robin, V.; Vignaud, L.; Toumi, N.; Cieslik, A.; Achiron, A.; Carasso, R.L.; Gurevich, M.; Braverman, I.; et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann. Neurol. 2012, 71, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.M.K.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.Y.E.; Trojanowski, J.Q.; Lee, V.M.Y.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.W.H.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.J.; Lubsen, N.H.; Kampinga, H.H. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 2010, 37, 355–369. [Google Scholar] [CrossRef]
- Scior, A.; Buntru, A.; Arnsburg, K.; Ast, A.; Iburg, M.; Juenemann, K.; Pigazzini, M.L.; Mlody, B.; Puchkov, D.; Priller, J.; et al. Complete suppression of Htt fibrillization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J. 2017, 37, 282–298. [Google Scholar] [CrossRef]
- Bersuker, K.; Hipp, M.S.; Calamini, B.; Morimoto, R.I.; Kopito, R.R. Heat shock response activation exacerbates inclusion body formation in a cellular model of Huntington disease. J. Biol. Chem. 2013, 288, 23633–23638. [Google Scholar] [CrossRef]
- Benson, M.D.; Uemichi, T. Transthyretin amyloidosis. Amyloid 1996, 3, 44–56. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Said, G. Transthyretin related familial amyloid polyneuropathy. Curr. Opin. Neurol. 2000, 13, 569–573. [Google Scholar] [CrossRef]
- Oroz, J.; Kim, J.H.; Chang, B.J.; Zweckstetter, M. Mechanistic basis for the recognition of a misfolded protein by the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 2017, 24, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oroz, J.; Zweckstetter, M. Structure of monomeric transthyretin carrying the clinically important T119M mutation. Angew. Chem. Int. Ed. 2016, 55, 16168–16171. [Google Scholar] [CrossRef]
- Santos, S.D.; Magalhães, J.; Saraiva, M.J. Activation of the heat shock response in familial amyloidotic polyneuropathy. J. Neuropathol. Exp. Neurol. 2008, 67, 449–455. [Google Scholar] [CrossRef]
- Masser, A.E.; Kang, W.; Roy, J.; Kaimal, J.M.; Quintana-Cordero, J.; Friedländer, M.R.; Andréasson, C. Cytoplasmic protein misfolding titrates Hsp70 to activate nuclear Hsf1. eLife 2019, 8, e47791. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Funez, P.; Casas-Tinto, S.; Zhang, Y.; Gómez-Velazquez, M.; Morales-Garza, M.A.; Cepeda-Nieto, A.C.; Castilla, J.; Soto, C.; Rincon-Limas, D.E. In vivo generation of neurotoxic prion protein: Role for Hsp70 in accumulation of misfolded isoforms. PLoS Genet. 2009, 5, e1000507. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Barbieri, L.; Rubino, J.T.; Kozyreva, T.; Cantini, F.; Banci, L. In-cell NMR reveals potential precursor of toxic species from SOD1 fALS mutants. Nat. Commun. 2014, 5, 5502. [Google Scholar] [CrossRef] [PubMed]
- Zetterström, P.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Marklund, S.L. Proteins that bind to misfolded mutant superoxide dismutase-1 in spinal cord from transgenic amyotrophic lateral sclerosis (ALS) model mice. J. Biol. Chem. 2011, 286, 20130–20136. [Google Scholar] [CrossRef] [PubMed]
- Claes, F.; Rudyak, S.; Laird, A.S.; Louros, N.; Beerten, J.; Debulpaep, M.; Michiels, E.; van der Kant, R.; Van Durme, J.; De Baets, G.; et al. Exposure of a cryptic Hsp70 binding site determines the cytotoxicity of the ALS-associated SOD1-mutant A4V. Protein Eng. Des. Sel. 2019, 32, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef]
- Perez-Canadillas, J.M.; Tidow, H.; Freund, S.M.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution structure of p53 core domain: Structural basis for its instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef]
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-mediated regulation of the conformation of p53 DNA binding domain and p53 cancer variants. Mol. Cell 2019, 74, 831–843. [Google Scholar] [CrossRef]
- Akakura, S.; Yoshida, M.; Yoneda, Y.; Horinouchi, S. A role for Hsc70 in regulating nucleocytoplasmic transport of a temperature-sensitive p53 (p53Val-135). J. Biol. Chem. 2001, 276, 14649–14657. [Google Scholar] [CrossRef]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jäättelä, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar] [CrossRef]
- Dahiya, V.; Agam, G.; Lawatscheck, J.; Rutz, D.A.; Lamb, D.C.; Buchner, J. Coordinated conformational processing of the tumor suppressor protein p53 by the Hsp70 and Hsp90 chaperone machineries. Mol. Cell 2019, 74, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Monsellier, E.; Redeker, V.; Ruiz-Arlandis, G.; Bousset, L.; Melki, R. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 2015, 290, 2560–2576. [Google Scholar] [CrossRef]
- Escobedo, A.; Topal, B.; Kunze, M.B.A.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairí, M.; Pierattelli, R.; et al. Side chain to main chain hydrogen bonds stabilize a polyglutamine helix in a transcription factor. Nat. Commun. 2019, 10, 2034. [Google Scholar] [CrossRef]
- Bongiovanni, M.N.; Aprile, F.A.; Sormanni, P.; Vendruscolo, M. A rationally designed Hsp70 variant rescues the aggregation-associated toxicity of human IAPP in cultured pancreatic islet β-cells. Int. J. Mol. Sci. 2018, 19, 1443. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Folorunso, O.; Taglialatela, G.; Pierce, A. Overexpression of heat shock factor 1 maintains TAR DNA binding protein 43 solubility via induction of inducible heat shock protein 70 in cultured cells. J. Neurosci. Res. 2016, 94, 671–682. [Google Scholar] [CrossRef]
- Udan-Johns, M.; Bengoechea, R.; Bell, S.; Shao, J.; Diamond, M.I.; True, H.L.; Weihl, C.C.; Baloh, R.H. Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 2014, 23, 157–170. [Google Scholar] [CrossRef]
- Park, S.J.; Borin, B.N.; Martinez-Yamout, M.A.; Dyson, H.J. The client protein p53 adopts a molten globule-like state in the presence of Hsp90. Nat. Struct. Mol. Biol. 2011, 18, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kostic, M.; Dyson, H.J. Dynamic interaction of Hsp90 with its client protein p53. J. Mol. Biol. 2011, 411, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, M.; Ozenne, V.; Bibow, S.; Jaremko, M.; Jaremko, L.; Gajda, M.; Jensen, M.R.; Biernat, J.; Becker, S.; Mandelkow, E.; et al. Predictive atomic resolution descriptions of intrinsically disordered htau40 and α-synuclein in solution from NMR and small angle scattering. Structure 2014, 22, 238–249. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A. Comparison of structure and dynamics of micelle-bound human α-synuclein and Pakinson disease variants. J. Biol. Chem. 2005, 280, 43179–43187. [Google Scholar] [CrossRef]
- He, W.-T.; Xue, W.; Gao, Y.-G.; Hong, J.-Y.; Yue, H.-W.; Jiang, L.-L.; Hu, H.-Y. HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci. Rep. 2017, 7, 14797. [Google Scholar] [CrossRef]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. α-synuclein regulation by chaperones in mammalian cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Soto, C.; Castaño, E.M.; Frangione, B.; Inestrosa, N.C. The α -helical to β-strand transition in the amino-terminal fragment of the amyloid β-peptide modulates amyloid formation. J. Biol. Chem. 1995, 270, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Abisambra, J.F.; Zhang, J.; Dharia, S.; O’Leary, J.C.; Patel, T.; Braswell, K.; Jani, T.; Gestwicki, J.E.; Dickey, C.A. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem. 2012, 287, 24814–24820. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Koren, J., 3rd; Dickey, C.A. Reconstructing the Hsp90/Tau machine. Curr. Enzym. Inhib. 2013, 9, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, M.; O’Leary, J.C., 3rd; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Baker, J.D.; Sabbagh, J.J.; Dickey, C.A. The emerging role of peptidyl-prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer’s disease. J. Neurochem. 2015, 133, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Laurents, D.V. RNA binding proteins: Diversity from microsurgeons to cowboys. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194398. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Handwerger, K.E.; Cordero, J.A.; Gall, J.G. Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol. Biol. Cell 2005, 16, 202–211. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef]
- Li, H.-R.; Chiang, W.-C.; Chou, P.-C.; Wang, W.-J.; Huang, J.-R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 2018, 293, 6090–6098. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.-M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 2018, 174, 688–699. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Hyman, A.A. Are aberrant phase transitions a driver of cellular aging? Bioessays 2016, 38, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Peskett, T.R.; Rau, F.; O’Driscoll, J.; Patani, R.; Saibil, H.R. A liquid to solid phase transition underlying pathological huntingtin exon1 aggregation. Mol. Cell 2018, 70, 588–601. [Google Scholar] [CrossRef]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 56, 508–513. [Google Scholar] [CrossRef]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.-E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef]
- Cherkasov, V.; Hofmann, S.; Druffel-Augustin, S.; Mogk, A.; Tyedmers, J.; Stoecklin, G.; Bukau, B. Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 2013, 23, 2452–2462. [Google Scholar] [CrossRef]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015, 4, e06807. [Google Scholar] [CrossRef]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Mateju, D.; Mediani, L.; Carra, S. Granulostasis: Protein quality control of RNP granules. Front. Mol. Neurosci. 2017, 10, 84. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef]
- Lechler, M.C.; Crawford, E.D.; Groh, N.; Widmaier, K.; Jung, R.; Kirstein, J.; Trinidad, J.C.; Burlingame, A.L.; David, D.C. Reduced insulin/IGF-1 signaling restores the dynamic properties of key stress granule proteins during aging. Cell Rep. 2017, 18, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Minami, M.; Shinozaki, F.; Suzuki, Y.; Abe, K.; Zenno, S.; Matsumoto, S.; Minami, Y. Hsp90 is involved in the formation of P-bodies and stress granules. Biochem. Biophys. Res. Comm. 2011, 407, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-Y.; Hou, S.-C.; Way, T.-D.; Wong, C.-H.; Wang, I.-F. Heat-shock protein dysregulation is associated with functional and pathological TDP-43 aggregation. Nat. Commun. 2013, 4, 2757. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Paine-Murrieta, G.D.; Taylor, C.W.; Pulcini, E.J.; Akinaga, S.; Benjamin, I.J.; Whitesell, L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer. Res. 2000, 6, 3312–3318. [Google Scholar]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef]
- Mediani, L.; Galli, V.; Carrà, A.D.; Bigi, I.; Vinet, J.; Ganassi, M.; Antoniani, F.; Tiago, T.; Cimino, M.; Mateju, D.; et al. BAG3 and BAG6 differentially affect the dynamics of stress granules by targeting distinct subsets of defective polypeptides released from ribosomes. Cell Stress Chaper. 2020, 25, 1045–1058. [Google Scholar] [CrossRef]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Audas, T.E.; Audas, D.E.; Jacob, M.D.; Ho, J.J.D.; Khacho, M.; Wang, M.; Perera, J.K.; Gardiner, C.; Bennett, C.A.; Head, T.; et al. Adaptation to stressors by systemic protein amyloidogenesis. Dev. Cell 2016, 39, 155–168. [Google Scholar] [CrossRef]



