 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tomar Ghansah | + 3863 word(s) | 3863 | 2020-12-10 07:12:30 | | | |
| 2 | Dean Liu | -1485 word(s) | 2378 | 2020-12-17 03:44:33 | | |
Video Upload Options
The bioflavonoid apigenin (API) is used to reduce inflammation in different PC models. Wild type mice harboring heterotopic or orthotopic PC were treated with API, which induced SHIP-1 expression, reduced inflammatory tumor-derived factors (TDF), increased the proportion of tumoricidal macrophages and enhanced anti-tumor immune responses, resulting in a reduction in tumor burden compared to vehicle-treated PC mice. In contrast, SHIP-1-deficient mice exhibited an increased tumor burden and displayed augmented proportions of pro-tumor macrophages. These results provide further support for the importance of SHIP-1 expression in promoting pro-tumor macrophage development in the pancreatic TME.
1. Introduction
Pancreatic cancer (PC) is one of the most aggressive and lethal cancers with a significantly higher mortality than other cancers[1]. Despite recent advances in cancer therapy, effective treatments for PC remain elusive. PC has heightened resistance to conventional treatments, such as immunotherapy and chemotherapy, in part, due to the inflammatory tumor microenvironment (TME) that contributes to the expansion of immunosuppressive myeloid-derived suppressor cells (MDSC) and Tumor-Associated Macrophages (TAM)[2][3]. These immunosuppressive cells specifically inhibit anti-tumor immune responses by halting the recruitment of effector immune cells to the TME, limiting the efficacy of current immunotherapies. In addition, MDSC and TAM interact with other immunosuppressive regulatory T cells (Tregs), immune cells, soluble factors, and other components in the stroma, which further exacerbates tumor progression, promotes metastasis and also chemoresistance[4][5][6]. Therefore, suppressing inflammation in the pancreatic TME may reduce the development of immunosuppressive MDSC and TAM and increase anti-tumor responses, thereby enhancing the efficacy of current therapies.
A chronic inflammatory TME is established through the production of pancreatic tumor-derived factors (TDF) (i.e., GM-CSF, IL-6, IFN-γ, and MCP-1) that induce the expansion and recruitment of MDSC from the bone marrow (BM) [7][8]. These MDSC consist of immature myeloid cells, macrophages, granulocytes and dendritic cells (DC) that aggressively suppress anti-tumor immunity[9][10]. MDSC encompass two different subsets, monocytic (M-MDSC) and granulocytic (G-MDSC); both have different modes of action in suppressing tumor immunity[11]. Mouse and human M-MDSC are known to be more suppressive than G-MDSC[10]. In fact, the production of monocyte chemoattractant protein-1 (MCP-1), as well as other inflammatory chemokines produced by the tumor, induce mobilization of M-MDSC into the TME, where they differentiate into immunosuppressive TAM, which have the potential to further suppress anti-tumor immune responses in PC[8][12][13][14].
TAM are macrophages with plasticity that can change phenotype to become either immunogenic or immunosuppressive in response to cytokines, chemokines and other soluble factors in the TME[8][15]. TAM can be either M1-like macrophages (M1 TAM) that are tumoricidal, or M2-like macrophages (M2 TAM) that are pro-tumor; both can be found in the TME of mice and humans[15]. However, the pancreatic TME strongly favors M-MDSC polarization into M2 TAM, which facilitates tumor progression, metastasis and chemoresistance [8][15]. Importantly, the disruption of inflammatory cytokine and chemokine pathways has been shown to decrease M-MDSC recruitment into the TME, correlating with tumor regression in both pre-clinical models and patients with PC [8][16]. Thus, modulating MDSC expansion or their mobilization to the TME present potentially promising strategies to constrain MDSC–TAM-associated immunosuppression in PC. Hence, the identification of new potential molecular target(s) or pathways that can regulate the development, function or mobilization of MDSC–TAM may increase anti-tumor immune responses.
Src Homology-2 (SH2) domain-containing Inositol 5′-Phosphatase-1 (SHIP-1) is a 145 kDa protein that regulates the activity of immune cells including myeloid cells, macrophages and DC[17][18]. SHIP-1 expression is differentially and developmentally regulated in immune cells by external soluble factors such as cytokines and chemokines in the microenvironment[19]. A key study reported that SHIP-1 expression is important for repressing macrophage polarization, with alveolar, peritoneal and BM-derived macrophages from SHIP-1 knockout (KO) mice exhibiting an M2-like macrophage phenotype[20]. SHIP-1 negatively regulates phosphatidylinositol 3-kinase (PI3K) activity in immune cells[17][21][22]. As a result, SHIP-1 regulates the activity of numerous signaling pathways, including those involved in cell differentiation, proliferation, apoptosis, mobilization and function of myeloid immune cells[17][23]. Deficiency in SHIP-1 expression results in chronic myeloid leukemia in both humans and mice[24], consistent with studies reporting that SHIP-1 acts as a tumor suppressor preventing metastasis in a pre-clinical lung cancer model [25]. In previous studies, we reported a significant expansion of immunosuppressive macrophages in SHIP-1-deficient mice[26]. In addition, we also reported that mice with PC, SHIP-1 protein levels were significantly downregulated[21], and there were changes in the MDSC compartment that corresponded with an increase in tumor burden[21]. Thus, we have established that SHIP-1 is essential for maintaining myeloid homeostasis and function and shown that dysregulation of SHIP-1 can promote a pro-tumor microenvironment. Therefore, induction of SHIP-1 expression may augment tumoricidal TAM and improve anti-tumor immune responses in PC.
Recently, natural compounds known as bioflavonoids have been explored for their anti-tumor, anti-chemoresistance and anti-inflammatory properties against different cancers [28][29][30][31]. The bioflavonoid apigenin (API) has demonstrated potent anti-tumor activity and the ability to reduce chemoresistance to gemcitabine (one of the chemotherapy drugs used for PC) in human PC cell lines[32]. Amongst the bioactive flavonoids, API showed the most selective killing of cancer cells (i.e., rapidly dividing cells) while sparing normal cells[33]. In human PC cell lines, API induces apoptosis, cell-cycle arrest and inhibits DNA synthesis[28][34][35]. API has also been assessed in breast, prostate and PC pre-clinical animal models [36][37]. Our research group recently reported that API reduced tumor burden, improved anti-tumor immune responses and increased survival rates of mice bearing pancreatic tumors compared to vehicle-treated mice with PC[37].
2. Role of API in Treatment of PC
One of the major barriers for PC treatment is the expansion and accumulation of immunosuppressive MDSC and TAM that inhibit effector immune cells mobilizing to the TME to eradicate the tumor[3][6]. Thus, identifying new molecular targets that regulate MDSC and TAM development is urgently needed to reduce immunosuppression and to improve the efficacy of current PC treatments. In this study, we are the first to show that SHIP-1 gene and protein expression is dampened in the setting of PC. In addition, we show that the anti-inflammatory drug API reduced inflammatory cytokines and chemokines in experimental PC models, which correlated with an increase in SHIP-1 expression. This restoration in SHIP-1 expression by API treatment significantly promoted the development of anti-tumor M1-like TAM in the TME of these pre-clinical models. More importantly, API treatment increased the mobilization of effector CD8+ T cells and augmented their anti-tumor activity in the TME of OPC mice. In addition, we show that pancreatic tumors grow more rapidly in SHIPKO mice and their TAM are skewed toward a pro-tumor phenotype (M2-like TAM). This suggests that SHIP-1 is an important regulator of macrophage skewing in the TME of PC. Our results support the notion that SHIP-1 controls the plasticity of macrophages, with its enhanced expression promoting immunogenic M1-like TAM over immunosuppressive M2-like TAM in the pancreatic TME. Therefore, amplification of SHIP-1 expression may be a novel means by which macrophages could be polarized towards an anti-tumor phenotype in the pancreatic TME for the treatment of PC.
SHIP-1 expression is important for appropriate regulation of hematopoiesis and the maturation of myeloid cells into granulocytes, macrophages, and DC (Figure 1). Loss of SHIP-1 expression impacts hematopoietic stem cells (HSC) and exacerbates altered myelopoiesis, which leads to the development of immunosuppressive MDSCs, Tregs, and pro-tumor M2-like TAM in the pancreatic TME, thus facilitating PC tumor progression (Figure 1). In this study, we showed that API treatment reduces pancreatic TDFs such as IL-6, IFN-γ, TNF-α and MCP-1, which in turn, restores SHIP-1 expression. This likely leads to an adjustment of myelopoiesis and the development of tumoricidal M1-like TAM in PC models (Figure 1). API is a bioflavonoid known to have multiple targets such as casein kinase II (CK2), microRNAs (miRNAs), cytokine and chemokine signaling pathways, and it is an inhibitor of proteasomal degradation [37][42]. Currently, we do not know the exact targets that induce transcriptional suppression of SHIP-1 in our PC models and we are performing additional experiments to identify these mediators.
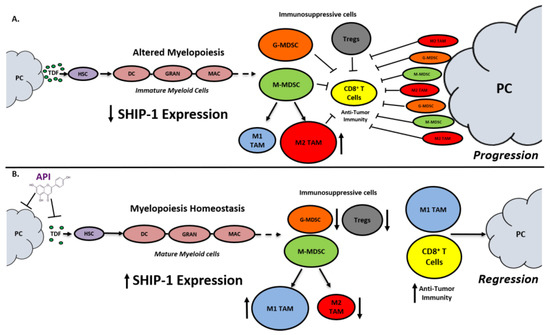
Figure 1. Proposed Model. Apigenin Increases SHIP-1 Expression, Augments Tumoricidal Macrophages and Improves Anti-Tumor Immune Responses in PC: (A) Pancreatic Tumor Microenvironment without Apigenin (API): Pancreatic cancer cells release tumor-derived factors (TDF) (green) that cause a decrease in SHIP-1 expression. The reduction in SHIP-1 expression causes hematopoietic stem cells (HSC) (purple) to skew towards altered myelopoiesis which yields immature myeloid cells (pink), including dendritic cells (DC), granulocytes (GRAN), and macrophages (MAC). This leads to the expansion of MDSC subsets, G-MDSC (orange) and M-MDSC (light green). M-MDSC mobilizes into the TME and differentiate into more pro-tumor M2-like TAM (red) compared to M1-like TAM (blue). MDSC subsets influence Treg (gray) expansion. MDSC subsets, M2-like TAM and Treg prevent anti-tumor immunity by blocking the migration of effector CD8+ T cells into the TME which leads to tumor progression. (B) Pancreatic Tumor Microenvironment with API: API targets and reduces the production of TDFs (directly or indirectly), leading to increased SHIP-1 expression, which promotes myelopoiesis homeostasis and the development of mature myeloid cells (DC, GRAN, and MAC). These M-MDSC that mobilize to the TME reprogram into tumoricidal M1-like TAM compared to M2-like TAM. Moreover, API treatment leads to a decrease in G-MDSC, M2-like TAM and Treg percentages in the TME. More importantly, API treatment increases the migration of effector CD8+ T cells (i.e., CD3+ TILs) into the TME, which elicits its anti-tumor activity, that causes pancreatic tumor regression.
G-MDSC can be transformed into Tumor-Associated Neutrophils (TAN) in the TME, but this phenomenon has been poorly investigated[43]. These TAN consist of N1-like TAN (N1 TAN) that are tumoricidal and N2-like TAN (N2 TAN) that have pro-tumor activity[43]. These TAN are recruited to the TME by inflammatory cytokines and chemokines and can only be distinguished due to their activation and cytokine production[43]. We observed that API treatment caused a significant reduction in the proportion of G-MDSC but no difference in M-MDSC in the TME of OPC and KC-HPC models. Interestingly, we observed a significant reduction in splenic G-MDSC percentages in CTRL-API (tumor-free) compared to vehicle-treated CTRL mice. It appears that API reduces G-MDSC percentages which is a new finding we are currently investigating. The phenotypic markers that distinguish N1 and N2 TAN in the TME of PC mice are unclear but may also include neutrophil chemokine receptors such as CXCR2[43]. Our studies implicate SHIP-1 expression as a contributor to G-MDSC homeostasis and the regulation of TAN (N1 vs. N2) in the TME that may influence PC progression and treatment resistance.
M-MDSC that mobilize to the TME develop into TAM[11]. However, we observed no differences in the proportion of M-MDSC vs. G-MDSC in the TME of API-treated OPC or KC-HPC mice, despite significant differences in M-MDSC and G-MDSC subsets in the spleen of HPC, OPC and KC-HPC mice treated with API. It has been reported that inflammatory monocytic-derived dendritic cells (inf-MoDC) are present in the TME, where they are phenotypically and physiologically distinct from M1 and M2 TAM[10][14]. It is known that SHIP-1 is important for DC development and function [18]. Therefore, we speculate that inf-MoDC are present and may have a dominant role in the TME of our OPC and KC-HPC models. This may explain why we did not observe any difference in a proportion of the M-MDSC subset in our models in the presence or absence of API. We have yet to fully immunophenotype inf-MoDC according to the current literature[14] or examine responses to API.
SHIP-1 expression is known to influence the development of immunosuppressive Treg and immunosuppressive TH17 immune cells in association with MDSC [9]. However, we did not evaluate TH17 percentages in our PC models. Yet, the mechanisms by which Treg and TH17 interplay with MDSC in the pancreatic TME treated with and without API warrants further investigation.
It is important to discuss Src homology 2 (SH-2) domain-containing tyrosine phosphatase-1 and -2 (SHP-1 and SHP-2) and their influence on PC progression[44][46]. SHP-1 acts as a tumor suppressor that potentially regulates immune checkpoint molecule expression, which can impact TIL mobilization into the TME of PC[45]. The overexpression of SHP-2 is considered to be biomarker and sign of poor prognosis for PC[46]. Therefore, SHP-2 acts as an oncogene that is important for Kras activation in human PC, which impacts signaling events that can control PC progression [47][48]. We reported in this study that API treatment of our OPC mice resulted in an increase in the mobilization of TILs into the TME that corresponded with a reduction in tumor burden. Moreover, we reported that API reduced PC induced inflammatory factors that are known to be potentially governed by downstream Kras signaling events (i.e., Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) cytokine-dependent signaling pathways). However, the mechanisms by which API regulates SHP-1/SHP-2 activity and signaling impacts PC outcomes warrant further investigation.
There are limitations in this study. First, a normal pancreas lacks infiltrating immune cells[49], therefore we were not able to evaluate its immune cell compartment even following enzymatic tissue digestion, in comparison to pancreatic tumors that are readily infiltrated by MDSCs, TAM and CD8+ T cells. Secondly, we were not able to yield enough pancreas protein lysates from CTRL mice (no tumor), which coincided with minimal SHIP-1 expression observed in our results from Western blot. Thirdly, API as a single agent therapy may be limited, however, we used API as an anti-inflammatory drug and as a tool to reduce tumor-induced inflammation. We found that API increased SHIP-1 expression, skewed macrophages to an anti-tumor phenotype and delayed tumor progression in our PC models. Therefore, in a clinical setting API may be used as an adjuvant with other PC therapeutic regimen. More importantly, we show here and previously published that API therapy (25 mg/kg) has no apparent side effects in our established PC models[37], which is consistent with its non-toxic properties in other pre-clinical models of cancer[50][51]. In fact, there are several other pre-clinical studies using higher dosages (i.e., 300 mg/kg and 50 µg/day) and longer durations of API treatments (up to 20 weeks) [52][53][54] In this study, we use API as a tool to increase SHIP-1 expression, the percentages of tumoricidal macrophages (M1-like TAM) and anti-tumor immunity in pre-clinical PC models. Taken together, our results suggest that SHIP-1 may be a potential and novel therapeutic target to be used as an interventional strategy for treating human pancreatic cancer.
References
- Mizrahi:, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020.
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506.
- Lafaro, K.J.; Melstrom, L.G. The paradoxical web of pancreatic cancer tumor microenvironment. Am. J. Pathol. 2019, 189, 44–57.
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419.
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527.
- Young, K.; Hughes, D.J.; Cunningham, D.; Starling, N. Immunotherapy and pancreatic cancer: Unique challenges and potential opportunities. Ther. Adv. Med Oncol. 2018, 10, 1758835918816281.
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835.
- Valilou, S.F.; Keshavarz-Fathi, M.; Silvestris, N.; Argentiero, A.; Rezaei, N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018, 39, 46–61.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174.
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220.
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8.
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550.
- Mielgo, A.; Schmid, M.C. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013, 46, 131–138.
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51.
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76.
- Cullinan, D.R.; Cripe, J.C.; Hawkins, W.; Goedegebuure, S. Radioprotective properties of myeloid-derived suppressor cells. Transl. Cancer Res. 2016, 5, S923–S925.
- Hamilton, M.J.; Ho, V.W.; Kuroda, E.; Ruschmann, J.; Antignano, F.; Lam, V.; Krystal, G. Role of SHIP in cancer. Exp. Hematol. 2011, 39, 2–13.
- Antignano, F.; Ibaraki, M.; Kim, C.; Ruschmann, J.; Zhang, A.; Helgason, C.D.; Krystal, G. SHIP is required for dendritic cell maturation. J. Immunol. 2010, 184, 2805–2813.
- Kalesnikoff, J.; Sly, L.M.; Hughes, M.R.; Buchse, T.; Rauh, M.J.; Cao, L.P.; Lam, V.; Mui, A.; Huber, M.; Krystal, G. The role of SHIP in cytokine-induced signaling. Rev. Physiol. Biochem. Pharmacol. 2003, 149, 87–103.
- Rauh, M.J.; Ho, V.; Pereira, C.; Sham, A.; Sly, L.M.; Lam, V.; Huxham, L.; Minchinton, A.I.; Mui, A.; Krystal, G. SHIP represses the generation of alternatively activated macrophages. Immunity 2005, 23, 361–374.
- Pilon-Thomas, S.; Nelson, N.; Vohra, N.; Jerald, M.; Pendleton, L.; Szekeres, K.; Ghansah, T. Murine pancreatic adenocarcinoma dampens SHIP-1 expression and alters MDSC homeostasis and function. PLoS ONE 2011, 6, e27729.
- Conde, C.; Gloire, G.; Piette, J. Enzymatic and non-enzymatic activities of SHIP-1 in signal transduction and cancer. Biochem. Pharmacol. 2011, 82, 1320–1334.
- Steelman, L.; Pohnert, S.; Shelton, J.; Franklin, R.; Bertrand, F.; McCubrey, J. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218.
- Lee, D.W.; Futami, M.; Carroll, M.; Feng, Y.; Wang, Z.; Fernandez, M.; Whichard, Z.; Chen, Y.; Kornblau, S.; Shpall, E.J.; et al. Loss of SHIP-1 protein expression in high-risk myelodysplastic syndromes is associated with miR-210 and miR-155. Oncogene 2012, 31, 4085–4094.
- Krystal, G.; Hamilton, M.J.; Bennewith, K.L. SHIP prevents metastasis. Aging (Albany NY) 2016, 8, 837–838.
- Ghansah, T.; Paraiso, K.H.; Highfill, S.; Desponts, C.; May, S.; McIntosh, J.K.; Wang, J.W.; Ninos, J.; Brayer, J.; Cheng, F.; et al. Expansion of myeloid suppressor cells in SHIP-deficient mice represses allogeneic T cell responses. J. Immunol. 2004, 173, 7324–7330.
- Ghansah, T. A novel strategy for modulation of MDSC to enhance cancer immunotherapy. Oncoimmunology 2012, 1, 984–985.
- Fendrich, V. Chemoprevention of pancreatic cancer—one step closer. Langenbeck’s Arch. Surg. 2012, 397, 495–505.
- Orfali, G.; Duarte, A.C.; Bonadio, V.; Martinez, N.P.; de Araujo, M.E.; Priviero, F.B.; Carvalho, P.O.; Priolli, D.G. Review of anticancer mechanisms of isoquercitin. World J. Clin. Oncol. 2016, 7, 189–199.
- Jiang, B.-H.; Qiu, J.-G.; Wang, L.; Liu, W.-J.; Wang, J.-F.; Zhao, E.-J.; Zhou, F.-M.; Ji, X.-B.; Wang, L.-H.; Xia, Z.-K. Apigenin inhibits IL-6 transcription and suppresses esophageal carcinogenesis. Front. Pharmacol. 2019, 10, 1002.
- Chen, X.J.; Wu, M.Y.; Li, D.H.; You, J. Apigenin inhibits glioma cell growth through promoting microRNA-16 and suppression of BCL-2 and nuclear factor-κB/MMP-9. Mol. Med. Rep. 2016, 14, 2352–2358.
- Johnson, J.L.; Gonzalez de Mejia, E. Interactions between dietary flavonoids apigenin or luteolin and chemotherapeutic drugs to potentiate anti-proliferative effect on human pancreatic cancer cells, in vitro. Food Chem. Toxicol. 2013, 60, 83–91.
- Shukla, S.; Gupta, S. Apigenin: A promising molecule for cancer prevention. Pharm. Res. 2010, 27, 962–978.
- Johnson, J.L.; de Mejia, E.G. Flavonoid apigenin modified gene expression associated with inflammation and cancer and induced apoptosis in human pancreatic cancer cells through inhibition of GSK-3beta/NF-kappaB signaling cascade. Mol. Nutr. Food Res. 2013, 57, 2112–2127.
- King, J.C.; Lu, Q.Y.; Li, G.; Moro, A.; Takahashi, H.; Chen, M.; Go, V.L.; Reber, H.A.; Eibl, G.; Hines, O.J. Evidence for activation of mutated p53 by apigenin in human pancreatic cancer. Biochim. Biophys. Acta 2012, 1823, 593–604.
- Madunić, J.; Madunić, I.V.; Gajski, G.; Popić, J.; Garaj-Vrhovac, V. Apigenin: A dietary flavonoid with diverse anticancer properties. Cancer Lett. 2018, 413, 11–22.
- Nelson, N.; Szekeres, K.; Iclozan, C.; Rivera, I.O.; McGill, A.; Johnson, G.; Nwogu, O.; Ghansah, T. Apigenin: Selective CK2 inhibitor increases Ikaros expression and improves T cell homeostasis and function in murine pancreatic cancer. PLoS ONE 2017, 12, e0170197.
- Shukla, S.; Gupta, S. Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radic. Biol. Med. 2008, 44, 1833–1845.
- Mukherjee, O.; Weingarten, L.; Padberg, I.; Pracht, C.; Sinha, R.; Hochdörfer, T.; Kuppig, S.; Backofen, R.; Reth, M.; Huber, M. The SH2-domain of SHIP1 interacts with the SHIP1 C-terminus: Impact on SHIP1/Ig-α interaction. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 206–214.
- Torres, M.P.; Rachagani, S.; Souchek, J.J.; Mallya, K.; Johansson, S.L.; Batra, S.K. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: Applications in diagnosis and therapy. PLoS ONE 2013, 8, e80580.
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332.
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50.
- Mukaida, N.; Sasaki, S.-i.; Baba, T. Two-Faced Roles of Tumor-Associated Neutrophils in Cancer Development and Progression. Int. J. Mol. Sci. 2020, 21, 3457.
- Collazo, M.M.; Wood, D.; Paraiso, K.H.; Lund, E.; Engelman, R.W.; Le, C.-T.; Stauch, D.; Kotsch, K.; Kerr, W.G. SHIP limits immunoregulatory capacity in the T-cell compartment. Blood 2009, 113, 2934–2944.
- Varone, A.; Spano, D.; Corda, D. Shp1 in solid cancers and their therapy. Front. Oncol. 2020, 10, 935.
- Zheng, J.; Huang, S.; Huang, Y.; Song, L.; Yin, Y.; Kong, W.; Chen, X.; Ouyang, X. Expression and prognosis value of SHP2 in patients with pancreatic ductal adenocarcinoma. Tumor Biol. 2016, 37, 7853–7859.
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1-and RAS-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073.
- Ruckert, M.T.; de Andrade, P.V.; Santos, V.S.; Silveira, V.S. Protein tyrosine phosphatases: Promising targets in pancreatic ductal adenocarcinoma. Cell. Mol. Life Sci. 2019, 76, 2571–2592.
- Saurer, L.; Reber, P.; Schaffner, T.; Büchler, M.W.; Buri, C.; Kappeler, A.; Walz, A.; Friess, H.; Mueller, C. Differential expression of chemokines in normal pancreas and in chronic pancreatitis. Gastroenterology 2000, 118, 356–367.
- Singh, P.; Mishra, S.K.; Noel, S.; Sharma, S.; Rath, S.K. Acute exposure of apigenin induces hepatotoxicity in Swiss mice. PLoS ONE 2012, 7, e31964.
- Caltagirone, S.; Rossi, C.; Poggi, A.; Ranelletti, F.O.; Natali, P.G.; Brunetti, M.; Aiello, F.B.; Piantelli, M. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int. J. Cancer 2000, 87, 595–600.
- Ai, X.-Y.; Qin, Y.; Liu, H.-J.; Cui, Z.-H.; Li, M.; Yang, J.-H.; Zhong, W.-L.; Liu, Y.-R.; Chen, S.; Sun, T. Apigenin inhibits colonic inflammation and tumorigenesis by suppressing STAT3-NF-κB signaling. Oncotarget 2017, 8, 100216.
- Shukla, S.; Bhaskaran, N.; Babcook, M.A.; Fu, P.; MacLennan, G.T.; Gupta, S. Apigenin inhibits prostate cancer progression in TRAMP mice via targeting PI3K/Akt/FoxO pathway. Carcinogenesis 2014, 35, 452–460.
- Zhao, L.; Wang, J.-L.; Liu, R.; Li, X.-X.; Li, J.-F.; Zhang, L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer’s disease mouse model. Molecules 2013, 18, 9949–9965.


