 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | arrigo de benedetti | + 4835 word(s) | 4835 | 2020-12-11 03:27:11 | | | |
| 2 | Dean Liu | -2257 word(s) | 2578 | 2020-12-14 03:42:18 | | | | |
| 3 | Dean Liu | Meta information modification | 2578 | 2020-12-17 03:24:51 | | |
Video Upload Options
Most prostate cancer (PCa) deaths result from progressive failure in standard androgen deprivation therapy (ADT), leading to metastatic castration-resistant PCa (mCRPC); however, the mechanism and key players leading to this are not fully understood. While studying the role of tousled-like kinase 1 (TLK1) and Never in Mitosis Gene A (NIMA)-related kinase 1 (NEK1) in a DNA damage response (DDR)-mediated cell cycle arrest in LNCaP cells treated with bicalutamide, the overexpression of wt-NEK1 resulted in a rapid conversion to androgen-independent (AI) growth, analogous to what has been observed when YAP1 is overexpressed. It's reported that overexpression of wt-NEK1 results in accumulation of YAP1, suggesting the existence of a TLK1>NEK1>YAP1 axis that leads to adaptation to AI growth. Further, YAP1 is co-immunoprecipitated with NEK1. Importantly, NEK1 was able to phosphorylate YAP1 on six residues in vitro, which researchers believe are important for stabilization of the protein, possibly by increasing its interaction with transcriptional partners. In fact, knockout (KO) of NEK1 in NT1 PCa cells resulted in a parallel decrease of YAP1 level and reduced expression of typical YAP-regulated target genes. In terms of cancer potential implications, the expression of NEK1 and YAP1 proteins was found to be increased and correlated in several cancers. These include PCa stages according to Gleason score, head and neck squamous cell carcinoma, and glioblastoma, suggesting that this co-regulation is imparted by increased YAP1 stability when NEK1 is overexpressed or activated by TLK1, and not through transcriptional co-expression. Researchers propose that the TLK1>NEK1>YAP1 axis is a key determinant for cancer progression, particularly during the process of androgen-sensitive to -independent conversion during progression to mCRPC.
1. Introduction
The founding member of the NIMA (never in mitosis gene A) family of protein kinases was originally identified in Aspergillus nidulans as a protein kinase essential for mitosis[1], and expression of a dominant-negative mutant of NIMA results in G2 arrest in vertebrate cells[2]. NIMA-related kinases (NEKs) have adapted to a variety of cellular functions in addition to mitosis[3]. In human cells, 11 NEKs were identified that are involved in several functions. For example, NEK2 is critical for centrosome duplication[3], whereas NEK6, 7, and 9 are regulators of the mitotic spindle and cytokinesis[4]. NEK1, NEK4, NEK8, NEK10, and NEK11 have been linked to the DNA damage response (DDR) and DNA repair pathways as well as ciliogenesis[3]. NEK1 mediates Chk1 activation likely by modulating the ATRIP/ATR interaction and activity [5], although this may be controversial[6]. NEK1 activity and relocalization to nuclei were reported to increase upon a variety of genotoxic stresses[5][7]. A defect in DNA repair in NEK1-deficient cells is suggested by the persistence of Double Strand Breaks (DSBs) after low-dose ionizing radiation (IR). NEK1-deficient cells fail to activate the checkpoint kinases Chk1 and Chk2, and fail to arrest properly at G1/S- or G2/M-phase checkpoints after DNA damage[8]. NEK1-deficient cells suffer major errors in mitotic chromosome segregation and cytokinesis, and become aneuploid[9]. Genomic instability is also manifested in NEK1+/− mice, which later in life develop lymphomas with a higher incidence than wild type littermates[9]. NEK1 is also known to negatively regulate apoptosis by phosphorylating VDAC1, regulating the closure of the anion channel of the mitochondrial membrane, which promotes survival of renal cell carcinoma[10][11][12]. Loss of function mutation of NEK1 leads to DNA damage accumulation in the motorneurons that may lead to several neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS)[13][14]. NEK1 is associated with primary cilia and centrosomes[15][16], which was reported to be implicated in the development of polycystic kidney disease (PKD) when there is a NEK1 deficiency [17]. However, the precise mechanism leading to PKD due to NEK1 insufficiency is not clear, but a clue came from the discovery that NEK1 interacts with and phosphorylates TAZ, involved in the E3 ligase complex, which regulates the stability of polycystin 2[18]. TAZ is also a paralog of yes-associated protein (YAP), a transcriptional coactivator that mediates many functions in normal development and in disease pathology, such as cancer progression, including prostate cancer [19][20][21][22].
We recently uncovered a new DDR axis involving the protein kinase tousled-like kinase1 (TLK1) as an early mediator of the DDR. TLK1 serves as an upstream activator of NEK1>ATR>Chk1[6][23], which has important implications during the early stages of prostate cancer (PCa) progression to androgen independence (AI) [24][25]. We found that overexpression of wt-NEK1 (but not the T141A kinase-hypoactive mutant that cannot be phosphorylated by TLK1) hastens the progression of LNCaP cells to androgen-independent growth[24]. The protective cell cycle arrest mediated by the TLK1>NEK1 DDR pathway seems insufficient to explain the rapid growth recovery observed in bicalutamide-treated cells when NEK1 is overexpressed, and suggests that NEK1 may have additional functions. We suspected that it may regulate the Hippo pathway, as it was reported that ectopic expression of YAP is sufficient to convert LNCaP cells from androgen-sensitive (AS) to AI in vitro[19]. NEK1 was also found to phosphorylate TAZ specifically at S309[18], and this was related to increased CTGF expression (one of TAZ/YAP transcriptional targets). TLKs may regulate the Hippo pathway through their activity on NEK1 upstream of YAP/TAZ. YAP/TAZ (60% identical) are the main effectors of the Hippo signaling pathway. This pathway is involved in regulating organ size through controlling multiple cellular functions including cell proliferation and apoptosis[26]. The Hippo pathway responds to a variety of signals, including cell–cell contact, mechano-transduction[21], and apico–basal polarity[20][26]. When the Hippo pathway is activated, kinases MST1/2 and LATS1/2 phosphorylate and inactivate YAP and TAZ. YAP and TAZ are transcriptional co-activators but lack DNA binding activity. Upon phosphorylation by MST and LATS kinases, they are sequestered in the cytoplasm, ubiquitylated by the β-TrCP ubiquitin ligase, and marked for proteasomal degradation (reviewed in[20]). YAP/TAZ are usually inhibited by cell–cell contact in normal tissues[26], while over-activation of YAP/TAZ through aberrant regulation of the Hippo pathway has been noted in many types of tumors. This is associated with the acquisition of malignant traits, including resistance to anticancer therapies; maintenance of cancer stem cells; distant metastasis[26]; and, in prostate, adenocarcinoma progression[27]. When the Hippo core kinases are “off”, YAP/TAZ translocate into the nucleus, binds to TEAD1–4, and activates the transcription of TEAD downstream target genes, leading to multiple oncogenic activities, including loss of contact inhibition, cell proliferation, epithelial–mesenchymal transition, and resistance to apoptosis. In PCa, YAP has been identified as an Androgen Receptor-binding partner that colocalizes with AR in both androgen-dependent and androgen-independent manners in castration-resistant PCa (CRPC) patients[27]. YAP is also found to be upregulated in AI-LNCaP-C4-2 cells and, when expressed ectopically in LNCaP cells, it activates AR signaling and confers castration resistance. Knockdown of YAP greatly reduces the rates of migration and invasion of LNCaP, and YAP-activated androgen receptor signaling is sufficient to promote LNCaP cells from an AS to an AI state in vitro, while YAP conferred castration resistance in vivo[19] It was also recently determined that ERG (and the common TMPRSS2–ERG fusion) activates the transcriptional program regulated by YAP1, and that prostate-specific activation of either ERG or YAP1 in mice induces similar transcriptional changes and results in age-related prostate tumors[28].
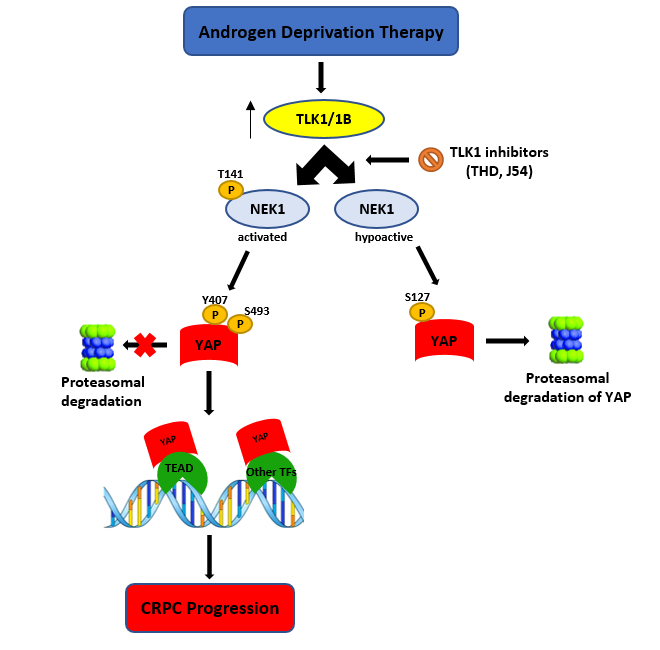
2. NEK1 Phosphorylation of YAP
During studies aimed at elucidating the process of ADT adaptation of AS PCa cell (initially in LNCaP), which proceeds through a process of activating the DDR and increased activity of the kinases TLK1B and NEK1 [11][24][25], we made the observation that overexpression of wt-NEK1, but not the hypoactive NEK1-T141A variant that cannot be activated by TLK, resulted in a rapid adaptation to bicalutamide and formation of AI colonies. From a review of the literature on the process of AI conversion of LNCaP and other studies of CRPC progression, we suspected the involvement of Hippo pathway deregulation and, in particular, YAP-driven gene expression (for a recent review, see[29]). Moreover, Yim et al. reported that NEK1 can phosphorylate TAZ and regulates its turnover rate[18]. Since YAP1 and TAZ are two highly homologous proteins that possess several conserved phospho-residues, we set out to investigate the protein level of YAP in LNCaP overexpressing wt-NEK1 and the T141A mutant in conjunction with a TLK inhibitor (THD) to suppress the activating phosphorylation of NEK1. Interestingly, we observed an increased degradation of YAP in cells overexpressing NEK1-T141A mutant or parental LNCaP treated with THD, in contrast to elevated level of YAP (and no degradation) in cells that overexpress wt-NEK1. Furthermore, treatment of LNCaP cells with THD resulted in downregulated expression of several YAP-dependent transcripts. As an indication that this is in fact a general phenomenon in PCa, increased degradation of YAP1 after inhibition of the TLK1>NEK1 axis with THD or J54 was independently verified in mouse NT1 cells. In addition, genetic depletion of NEK1 resulted in YAP1 loss and YAP1 target gene downregulation in NT1 cells. It should be noted that YAP is a generally unstable protein whose turnover rate is strongly regulated by multiple stabilizing[30] or de-stabilizing phosphorylation events controlled by multiple kinases (see[19][20][26]for some reviews). Large tumor suppressor 1 and 2 (LATS1/2), the core kinases of the Hippo signaling pathway, can phosphorylate YAP1 on Ser127 residue, which creates a binding site for 14-3-3 proteins. The 14-3-3 binding of YAP leads to the cytoplasmic sequestration of YAP[31][32]. Sequential phosphorylation by LATS1/2 on YAP Ser397 primes it for further phosphorylation by Casein Kinase CK1δ/ε on Ser400 and Ser403, which creates a phosphodegron motif for (Skp Cullin F box) β-TrCP/SCF E3 ubiquitin ligase-mediated proteasomal degradation[33]. Recent findings also identify factors such as NR4A1 (nuclear receptor superfamily) that regulate the 14-3-3 interaction with YAP1 and promote its ubiquitination and degradation[34]. Several other kinases independent of the Hippo pathway can regulate the stability of YAP1 protein. For instance, nuclear Dbf2-related kinase (NDR1/2) can also phosphorylate YAP on Ser127 residue and can promote its cytoplasmic retention, thereby negatively regulating YAP stability [35]. Evidence suggests that the protein kinase B/AKT can also phosphorylate YAP on Ser127 residue, leading to binding of 14-3-3 and cytoplasmic retention[31]. In contrast, several members of the Src family of kinases such as Src, Yes, and c-Abl can positively regulate YAP stability. c-Abl/Src/Yes are known to phosphorylate YAP on Tyr357 residue, which results in the nuclear translocation and, hence, stabilization of YAP[30][36][37]. Moreover, Ras-associated factor isoform 1C (RASSF1C) is known to promote tyrosine phosphorylation of YAP1 (Tyr357) through activated Src (pTyr416) and cause nuclear localization of YAP1[38]. Similarly, mitogen-activated protein kinases such as c-Jun-N-terminal kinases (JNK1/2) are also reported to be YAP kinases that phosphorylate YAP on Ser317 and Thr362, promoting YAP nuclear translocation and stabilization[39]. Thus, post-translational modifications such as phosphorylation determine YAP turnover rate and activity.
Therefore, we propose the phosphorylation of Y407 by Nek1 as one potential mechanism of YAP stabilization and increased transcriptional output, although the other 5-phosphorylation sites could be equally important. There are examples in YAP and TAZ where phosphorylation of some residues impairs ubiquitination and subsequent proteasomal degradation, as in one example, phosphorylation of S128 by NLK competed for the destabilizing LATS1-dependent S127 phosphorylation[40]. However, we currently favor a pY407-related mechanism based on the equivalent pY316 of TAZ, where it was shown that the phosphorylation of that residue, reportedly by c-Abl, was necessary to mediate its interaction with the transcription factor NFAT5[41]. This was implicated in an inhibitory pathway of NFAT5—a major osmoregulatory transcription factor—during hyperosmotic stress. Similarly, JNK1/2-mediated phosphorylation of YAP1 on Ser317 and Thr362 promotes YAP’s ability to bind and stabilize both pro-apoptotic p73 and pro-proliferative ΔNp63α in different cell types[39][42]. We think that, likewise, pY407 promotes the interaction of YAP with some of its transcriptional partners, and hence promotes its nuclear translocation, function, and stabilization, away from cytoplasmic degradation. Importantly, while the phosphorylation of Y407 was identified in proteomic studies[43], to our knowledge, the kinase responsible for it has not been reported.
Resistance to androgen deprivation therapy (ADT) promotes androgen-independent growth and proliferation of PCa cells, which requires efficient DNA damage response (DDR) and repair mechanisms, activation of compensatory signaling pathways, transcription factors, and co-factors to drive castration resistance. Findings from our lab and others suggest that ADT activates the TLK1-NEK1 signaling pathway that promotes PCa progression by activating the DDR[11][24]. Hyper-activation of NEK1 may also lengthen G2/M checkpoints, which provides the cells sufficient time to repair their damaged DNA after ADT or radiation therapy[7][44]. However, DDR alone may not be able to induce androgen-insensitive growth of PCa cells. Thus, we hypothesize that TLK1-NEK1 may be implicated in some other signaling pathway, leading to AI growth. YAP1 is a major oncoprotein that drives many different types of malignancies, including PCa[45], head and neck cancer[45], gastric cancer[46] colon cancer[46], thyroid cancer [47], lung cancer[48], ovarian cancer[49], and liver cancer [50]. NEK1-mediated phosphorylation of YAP1 (most probably on Tyr407 and/or Thr493) may induce a conformational change that counteracts the sequential phosphorylation by LATS1/2 and CK1δ/ε and subsequently protects YAP from proteasomal degradation. Moreover, Tyr407 lies on the transcriptional activation domain of YAP1, which may increase its interaction affinity to its assigned transcriptional factors[51]. Ectopic YAP expression was reported to drive LNCaP cells from androgen-sensitive to androgen-insensitive states[19]. Reducing the turnover rate will increase cellular accumulation of YAP, which can enable its oncogenic properties to drive castration resistance by several mechanisms. Previous studies reported that YAP can mediate PI (3)K-mTOR signaling and activate AKT[52][53][54]. Activation of mTOR will lead to enhanced translation of TLK1B that can, in turn, increase YAP1 phosphorylation through TLK1-NEK1 nexus. This suggests a positive feed-forward mechanism for YAP accumulation. Elevated YAP can also activate ERK that will promote cell proliferation in absence of AR signaling. Kuser-Abali et al. reported that AR and YAP can interact, and this interaction contributes to the switch from androgen-dependent to castration-resistant phenotype[55]. Overexpression of YAP can also regulate the expression of AR target genes, including PSA, NKX3.1, PGC-1, and KLK2, which suggests that YAP may control AR activity. YAP Tyr407 phosphorylation could increase the binding affinity of AR and AR ligand-insensitive variant AR-V7, thus contributing to androgen refractory growth of PCa cells. Therapy-induced YAP overexpression may also induce EMT activation by upregulating EMT-specific genes. Increasing the stemness of PCa cells can be another mechanism by which stabilized YAP can promote castration-resistant growth of PCa cells, which will further contribute to chemo-resistance of cancer cells [56]. Our bioinformatics analyses also suggested a link between NEK1 and YAP1 in different cancers. YAP1 protein level is abundant in high-grade PCa tumors, despite the progressive downregulation of YAP1 mRNA expression. Other groups also reported that YAP protein is positively correlated with the Gleason score, consistent with the findings of our bioinformatics analysis[57].
3. Conclusions
YAP’s transcriptional activity and degradation is mainly regulated by phosphorylation through several kinases dependent and independent of the Hippo pathway. Using small molecule inhibitors against YAP cannot completely abolish YAP transcriptional activity and is not very effective in treating YAP-driven cancers. Inhibitors such as verteporfin that can disrupt the YAP–TEAD interaction, but still cannot result in complete inhibition, as YAP can bind with other transcription factors such as TEF, SMADs, or TBX5. The majority of YAP kinases negatively regulate YAP by promoting its nuclear egress or degradation; however, NEK1 is found to stabilize YAP protein by phosphorylating it on several residues. Thus, targeting NEK1 or the TLK1–NEK1 axis can bring about therapeutic benefits in the clinical management of YAP-driven malignancies.x
References
- Osmani, S.A.; Pu, R.T.; Morris, N.R. Mitotic induction and maintenance by overexpression of a G2-specific gene that encodes a potential protein kinase. Cell 1988, 53, 237–244.
- Lu, K.P.; Hunter, T. Evidence for a NIMA-like mitotic pathway in vertebrate cells. Cell 1995, 81, 413–424, doi:410.1016/0092-8674(1095)90394-90391.
- Meirelles, G.V.; Perez, A.M.; de Souza, E.E.; Basei, F.L.; Papa, P.F.; Melo Hanchuk, T.D.; Cardoso, V.B.; Kobarg, J. "Stop Ne(c)king around": How interactomics contributes to functionally characterize Nek family kinases. World J. Biol. Chem. 2014, 5, 141–160.
- Moniz, L.; Dutt, P.; Haider, N.; Stambolic, V. Nek family of kinases in cell cycle, checkpoint control and cancer. Cell Div. 2011, 6, 18.
- Chen, Y.; Chen, C.F.; Riley, D.J.; Chen, P.L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655–663.
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180.
- Chen, Y.; Chen, P.L.; Chen, C.F.; Jiang, X.; Riley, D.J. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008, 7, 3194–3201.
- Pelegrini, A.L.; Moura, D.J.; Brenner, B.L.; Ledur, P.F.; Maques, G.P.; Henriques, J.A.; Saffi, J.; Lenz, G. Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis 2010, 25, 447–454.
- Chen, Y.; Chen, C.F.; Chiang, H.C.; Pena, M.; Polci, R.; Wei, R.L.; Edwards, R.A.; Hansel, D.E.; Chen, P.L.; Riley, D.J. Mutation of NIMA-related kinase 1 (NEK1) leads to chromosome instability. Mol. Cancer 2011, 10, 5.
- Chen Y, Gaczynska M, Osmulski P, Polci R, Riley DJ: Phosphorylation by Nek1 regulates opening and closing of voltage dependent anion channel 1. Biochem. Biophys. Res. Commun. 2010, 394, 798–803, doi:710.1016/j.bbrc.2010.1003.1077. Epub 2010 Mar 1015.
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 9, 1–13.
- Chen, Y.; Chen, C.F.; Polci, R.; Wei, R.; Riley, D.J.; Chen, P.L. Increased Nek1 expression in renal cell carcinoma cells is associated with decreased sensitivity to DNA-damaging treatment. Oncotarget 2014, 5, 4283–4294, doi:4210.18632/oncotarget.12005.
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150-162, doi:10.1016/j.scr.2018.1006.1005.
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Yoshimura, J.; Doi, K.; Morishita, S.; Goto, J.; Toda, T.; et al. Loss-of-function variants in NEK1 are associated with an increased risk of sporadic ALS in the Japanese population. J. Hum. Genet. 2020, 12, 020–00830.
- Mahjoub, M.R.; Trapp, M.L.; Quarmby, L.M. NIMA-related kinases defective in murine models of polycystic kidney diseases localize to primary cilia and centrosomes. J. Am. Soc. Nephrol. 2005, 16, 3485–3489, doi:3410.1681/ASN.2005080824.
- Shalom, O.; Shalva, N.; Altschuler, Y.; Motro, B.: The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 2008, 582, 1465–1470, doi:1410.1016/j.febslet.2008.1403.1036. Epub 2008 Apr 1461.
- Surpili, M.J.; Delben, T.M.; Kobarg, J. Identification of proteins that interact with the central coiled-coil region of the human protein kinase NEK1. Biochemistry 2003, 42, 15369–15376, doi:15310.11021/bi034575v.
- Yim, H.; Sung, C.K.; You, J.; Tian, Y.; Benjamin, T. Nek1 and TAZ interact to maintain normal levels of polycystin 2. J. Am. Soc. Nephrol. JASN 2011, 22, 832–837.
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell. Biol. 2015, 35, 1350–1362.
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874, doi:810.1101/gad.1909210.
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269, doi:210.1038/s41586-41018-40658-41581.
- Cheng, S.; Prieto-Dominguez, N.; Yang, S.; Connelly, Z.M.; StPierre, S.; Rushing, B.; Watkins, A.; Shi, L.; Lakey, M.; Baiamonte, L.B.; et al. The expression of YAP1 is increased in high-grade prostatic adenocarcinoma but is reduced in neuroendocrine prostate cancer. Prostate Cancer Prostatic Dis. 2020, 20, 020–0229.
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926, doi:910.1080/15384101.15382017.11314421. Epub 15382017 Apr 15384120.
- Singh, V.; Jaiswal, P.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of Androgen Independent Prostate tumors. Int. J. Cancer 2019, 145, 1055–1067.
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019, 453, 131–141. DOI 110.1016/j.canlet.2019.1003.1041.
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828, doi:810.1016/j.cell.2015.1010.1044.
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126–8126.
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808, doi:710.1016/j.ccell.2015.1005.1005.
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8, 370.
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361, doi:310.1016/j.molcel.2007.1012.1022.
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J.: Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003, 11, 11–23, doi:10.1016/s1097-2765(1002)00776-00771.
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312, doi:1210.1152/physrev.00005.02014.
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85, doi:10.1101/gad.184381
- He, L.; Yuan, L.; Yu, W.; Sun, Y.; Jiang, D.; Wang, X.; Feng, X.; Wang, Z.; Xu, J.; Yang, R.; et al. A Regulation Loop between YAP and NR4A1 Balances Cell Proliferation and Apoptosis. Cell Rep. 2020, 33, 108284.
- Zhang, L.; Tang, F.; Terracciano, L.; Hynx, D.; Kohler, R.; Bichet, S.; Hess, D.; Cron, P.; Hemmings, B.A.; Hergovich, A.; et al. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr. Biol. 2015, 25, 296–305, doi:210.1016/j.cub.2014.1011.1054.
- Li, B.; He, J.; Lv, H.; Liu, Y.; Lv, X.; Zhang, C.; Zhu, Y.; Ai, D. C-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J. Clin. Investig. 2019, 129, 1167–1179, doi:1110.1172/JCI122440. Epub 122019 Feb 122411.
- Sugihara, T.; Werneburg, N.W.; Hernandez, M.C.; Yang, L.; Kabashima, A.; Hirsova, P.; Yohanathan, L.; Sosa, C.; Truty, M.J.; Vasmatzis, G.; et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Mol. Cancer Res. 2018, 16, 1556–1567, doi:1510.1158/1541-7786.MCR-1518-0158.
- Vlahov, N.; Scrace, S.; Soto Manuel, S.; Grawenda Anna, M.; Bradley, L.; Pankova, D.; Papaspyropoulos, A.; Yee Karen, S.; Buffa, F.; Goding Colin, R.; et al. Alternate RASSF1 Transcripts Control SRC Activity, E-Cadherin Contacts, and YAP-Mediated Invasion. Curr. Biol. 2015, 25, 3019–3034.
- Tomlinson, V.; Gudmundsdottir, K.; Luong, P.; Leung, K.Y.; Knebel, A.; Basu, S. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 2010, 1, e29, doi:10.1038/cddis.2010.1037.
- Moon, S.; Kim, W.; Kim, S.; Kim, Y.; Song, Y.; Bilousov, O.; Kim, J.; Lee, T.; Cha, B.; Kim, M.; et al. Phosphorylation by NLK inhibits YAP-14-3-3-interactions and induces its nuclear localization. EMBO Rep. 2017, 18, 61–71.
- Jang, E.J.; Jeong, H.; Han, K.H.; Kwon, H.M.; Hong, J.H.; Hwang, E.S. TAZ suppresses NFAT5 activity through tyrosine phosphorylation. Mol. Cell. Biol. 2012, 32, 4925–4932, doi:4910.1128/MCB.00392-00312.
- Danovi, S.A.; Rossi, M.; Gudmundsdottir, K.; Yuan, M.; Melino, G.; Basu, S. Yes-associated protein (YAP) is a critical mediator of c-Jun-dependent apoptosis. Cell Death Differ. 2008, 15, 217–219, doi:210.1038/sj.cdd.4402226.
- Iliuk, A.B.; Martin, V.A.; Alicie, B.M.; Geahlen, R.L.; Tao, W.A. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell Proteom. 2010, 9, 2162–2172, doi:2110.1074/mcp.M2110.000091.
- Freund, I.; Hehlgans, S.; Martin, D.; Ensminger, M.; Fokas, E.; Rödel, C.; Löbrich, M.; Rödel, F. Fractionation-Dependent Radiosensitization by Molecular Targeting of Nek1. Cells 2020, 9, 1235, doi:1210.3390/cells9051235.
- Jiang, N.; Hjorth-Jensen, K.; Hekmat, O.; Iglesias-Gato, D.; Kruse, T.; Wang, C.; Wei, W.; Ke, B.; Yan, B.; Niu, Y.; et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene 2015, 34, 2764–2776, doi:2710.1038/onc.2014.2206.
- Kang, W.; Tong, J.H.; Chan, A.W.; Lee, T.L.; Lung, R.W.; Leung, P.P.; So, K.K.; Wu, K.; Fan, D.; Yu, J.; et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin. Cancer Res. 2011, 17, 2130–2139, doi:2110.1158/1078-0432.CCR-2110-2467.
- Lee, S.E.; Lee, J.U.; Lee, M.H.; Ryu, M.J.; Kim, S.J.; Kim, Y.K.; Choi, M.J.; Kim, K.S.; Kim, J.M.; Kim, J.W.; et al. RAF kinase inhibitor-independent constitutive activation of Yes-associated protein 1 promotes tumor progression in thyroid cancer. Oncogenesis 2013, 2, e55, doi:10.1038/oncsis.2013.1012.
- Xu, C.M.; Liu, W.W.; Liu, C.J.; Wen, C.; Lu, H.F.; Wan, F.S. Mst1 overexpression inhibited the growth of human non-small cell lung cancer in vitro and in vivo. Cancer Gene Ther. 2013, 20, 453–460, doi:410.1038/cgt.2013.1040.
- Steinhardt, A.A.; Gayyed, M.F.; Klein, A.P.; Dong, J.; Maitra, A.; Pan, D.; Montgomery, E.A.; Anders, R.A.: Expression of Yes-associated protein in common solid tumors. Hum. Pathol. 2008, 39, 1582–1589, doi:1510.1016/j.humpath.2008.1504.1012.
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009, 16, 425–438, doi:410.1016/j.ccr.2009.1009.1026.
- Yu, Y.; Su, X.; Qin, Q.; Hou, Y.; Zhang, X.; Zhang, H.; Jia, M.; Chen, Y. Yes-associated protein and transcriptional coactivator with PDZ-binding motif as new targets in cardiovascular diseases. Pharmacol. Res. 2020, 159, 105009, doi:10.1016/j.phrs.2020.105009.
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol. 2012, 14, 1322–1329, doi:1310.1038/ncb2615.
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 2011, 4, ra70, doi:10.1126/scisignal.2002278.
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240, doi:1210.1038/onc.2010.1504.
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126–8126
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1α. Exp. Cell Res. 2018, 371, 63–71, doi:10.1016/j.yexcr.2018.1007.1041. Epub 2018 Jul 1026.
- Sheng, X.; Li, W.B.; Wang, D.L.; Chen, K.H.; Cao, J.J.; Luo, Z.; He, J.; Li, M.C.; Liu, W.J.; Yu, C. YAP is closely correlated with castration-resistant prostate cancer, and downregulation of YAP reduces proliferation and induces apoptosis of PC-3 cells. Mol. Med. Rep. 2015, 12, 4867–4876, doi:4810.3892/mmr.2015.4005.


